


Team bringer subatomær opløsning til beregningsmikroskop
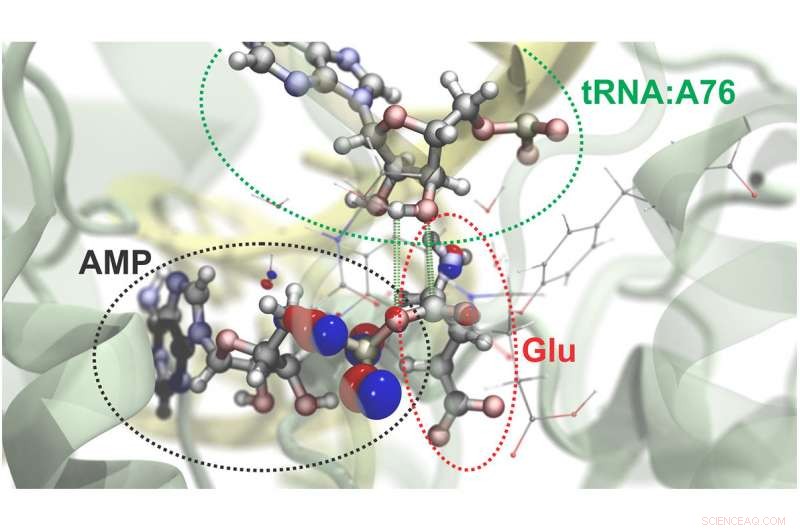
Forskere kan simulere atomisk og subatomær dynamik i store molekylære systemer. Her er en visualisering af den proces, ved hvilken aminosyren glutamat (Glu) er knyttet til en bestemt region af dets overførsels -RNA (tRNA). Et energirigt molekyle, ATP, driver denne reaktion og konverteres til AMP i processen. De røde og blå bobler repræsenterer sandsynligheden for at finde elektroner i bestemte områder. Grønne stiplede linjer afgrænser de atomer, der bindes i denne kemiske reaktion. Kredit:Rafael Bernardi, Zan Luthey-Schulten og Marcelo Melo
Forskere har bygget et "beregningsmikroskop", der kan simulere de atomare og subatomære kræfter, der driver molekylære interaktioner. Dette værktøj vil strømline indsatsen for at forstå livets kemi, model store molekylære systemer og udvikle nye farmaceutiske og industrielle midler, siger forskerne.
De rapporterer deres fund i journalen Naturmetoder .
Forskerne kombinerede to beregningsmetoder, der blev brugt til at simulere molekylære interaktioner. Den første, et nanoskala molekylær-dynamisk program kendt som NAMD, bruger klassisk-mekaniske metoder til at modellere strukturen og simulere adfærden hos hundredvis af millioner af individuelle atomer. Det andet program zoomer ind på det subatomære område, simulerer interaktioner mellem protoner, neutroner og elektroner. Modellering i denne kvantemekaniske skala kræver meget beregningskraft, så implementerede forskerne en metode til at opdele store molekyler i klassiske og kvantemekaniske områder. Dette giver dem mulighed for at fokusere deres beregningsressourcer på små regioner, der er involveret i kritiske interaktioner, såsom fremstilling eller afbrydelse af kemiske bindinger.
Både molekylærmekanik og kvantemekanik programmer har været tilgængelige i årevis, og andre teams har arbejdet på at kombinere dem, sagde University of Illinois kemiprofessor Zaida (Zan) Luthey-Schulten, der ledede den nye forskning sammen med sin mand, U. af I. fysikprofessor Klaus Schulten. Men den nye indsats effektiviserer etableringsprocessen, udføre og analysere simuleringerne.
"Vi konfigurerede det, så forskere let kan vælge, hvordan de vil opdele deres egne systemer, "Sagde Luthey-Schulten." Mine egne elever prøver det, og de fleste af dem er i stand til at gøre det uden store vanskeligheder. "
Schulten udviklede NAMD i Illinois i 1995, kombinerer det med en visualiseringssoftware, VMD, som gør det muligt for forskere at se store molekylære interaktioner udfolde sig. Schulten, der døde i 2016, sidestillede denne tilgang med at "bygge et beregningsmikroskop."
Det beregningsmikroskop er ideelt til modellering af strukturelle træk og bevægelser af store komplekser. For eksempel, i 2013, Schulten og hans kolleger brugte NAMD til at modellere HIV -kapsidet, som består af mere end 1, 300 identiske proteiner, der samles til en cagelike struktur, der beskytter virussen, indtil den kommer ind i en værtscelle. Denne simulering tegnede sig for interaktionerne mellem mere end 64 millioner atomer og krævede brug af Blue Waters -supercomputeren på National Center for Supercomputing Applications i U. I. Den nye undersøgelse gjorde også brug af Blue Waters, denne gang for at forbedre opløsningen af det beregningsmikroskop.

Fra venstre, kandidatstuderende Marcelo Melo, kemiprofessor Zaida Luthey-Schulten, postdoktorforsker Rafael Bernardi og deres kolleger har udviklet en ny tilgang til modellering af store molekylære interaktioner på atomare og subatomære skalaer. Deres arbejde strømliner metoden for andre forskere og studerende. Kredit:L. Brian Stauffer
NAMD -softwaren er designet til at beskrive adfærden hos individuelle atomer. Men individuelle atomer, der er involveret i specifikke kemiske interaktioner og reaktioner, opfører sig ikke altid som deres kolleger andre steder. At forstå, hvordan de varierer, kræver et nærmere kig på de subatomære kræfter, der spiller. Dette er især vigtigt i de dynamiske områder af molekyler - f.eks. de steder, hvor kemiske bindinger dannes eller brydes, sagde forskerne.
I den nye undersøgelse, forskergruppen i Illinois gik sammen med QM -eksperter Frank Neese, fra Max Planck Institute for Coal Research i Mulheim an der Ruhr, Tyskland; og Gerd B. Rocha, fra Federal University of Paraiba, i Joao Pessoa, Brasilien.
Som en demonstration af den nye tilgang, forskerne simulerede den kemiske adfærd af transfer -RNA'er, molekyler, der spiller en nøglerolle i oversættelsen af genetisk information til proteiner. Ved hjælp af NAMD, de modellerede den overordnede molekylære struktur af tRNA i det øjeblik, at et specielt protein indlæser en aminosyre til tRNA. De opdelte to steder i komplekset i regioner, der kræver den mere fokuserede kvantemekaniske tilgang. (Se en film af simuleringen.)
De subatomære simuleringer af interaktionerne mellem de to regioner tillod teamet at køre simuleringer af fire forskellige scenarier, der ville tillade tRNA at fungere, som det gør i cellen. Deres simuleringer afslørede, at en af de fire potentielle kemiske veje var mere energisk gunstig end de andre og dermed mere tilbøjelige til at forekomme.
Forskerne brugte også forskellige metoder til at opdele tRNA -komplekset mellem MM- og QM -regionerne og rapporterede om hver tilgang.
"Vi valgte ikke bare én måde; vi valgte så mange som muligt. Vi giver brugeren frihed. Hvordan du strukturerer det afhænger virkelig af det særlige system, du studerer, "sagde U. af I. postdoktorforsker Rafael Bernardi, en medlederforfatter på undersøgelsen med kandidatstuderende Marcelo Melo.
"Vi gør ikke hele systemet kvantemekanisk, fordi det ville tage evigt at beregne, "Sagde Melo.
"NAMD blev designet - og det var min mands vision - til at behandle virkelig store systemer, "Sagde Luthey-Schulten." Nu kan vi tilføje den subatomære skala til det, åbner op for store nye muligheder for forskning. "
 Varme artikler
Varme artikler
-
 1N4007 DiodespecifikationerEn ensretterdiode bruges som envejs kontrolventil. Da disse dioder kun tillader elektrisk strøm at strømme i en retning, bruges de til at konvertere vekselstrøm til jævnstrøm. Når du konstruerer en
1N4007 DiodespecifikationerEn ensretterdiode bruges som envejs kontrolventil. Da disse dioder kun tillader elektrisk strøm at strømme i en retning, bruges de til at konvertere vekselstrøm til jævnstrøm. Når du konstruerer en -
 Metode bruger radiosignaler til at afbilde skjulte objekter, der kører hurtigereIllustration af laboratorieopsætningen til m-Widar, med sendere og modtager til venstre og person bag wallboard til højre. Indsat nederst til højre viser det tilsvarende billede produceret af instrume
Metode bruger radiosignaler til at afbilde skjulte objekter, der kører hurtigereIllustration af laboratorieopsætningen til m-Widar, med sendere og modtager til venstre og person bag wallboard til højre. Indsat nederst til højre viser det tilsvarende billede produceret af instrume -
 En million pulser i sekundet:Hvordan partikelacceleratorer driver røntgenlasereSLAC National Accelerator Laboratory opgraderer sin Linac kohærente lyskilde, en røntgenlaser, at være et mere kraftfuldt værktøj til videnskab. Både Fermilab og Thomas Jefferson National Accelerator
En million pulser i sekundet:Hvordan partikelacceleratorer driver røntgenlasereSLAC National Accelerator Laboratory opgraderer sin Linac kohærente lyskilde, en røntgenlaser, at være et mere kraftfuldt værktøj til videnskab. Både Fermilab og Thomas Jefferson National Accelerator -
 Unik polymerbaseret fremstillingsproces til lave omkostninger, højere udbytte omprogrammerbare foto…Kredit:Eindhoven University of Technology Fremtiden ser lys ud for fotoniske integrerede kredsløb (PICer), da de ser ud til at blive brugt i kvantecomputere og deep learning-teknologier. Da PICer
Unik polymerbaseret fremstillingsproces til lave omkostninger, højere udbytte omprogrammerbare foto…Kredit:Eindhoven University of Technology Fremtiden ser lys ud for fotoniske integrerede kredsløb (PICer), da de ser ud til at blive brugt i kvantecomputere og deep learning-teknologier. Da PICer
- Brug af kunstig intelligens-værktøjer til at identificere niveauer af vold i film
- Undersøgelse beskriver, hvordan genomets tredimensionelle arkitektur ændres under cellecyklussen
- Forskere udvikler automatiseret robotudstyr til hurtigere blodprøver
- InSight-mission:Mars afsløret
- Anvendelsen af volumetrisk analyse
- Hvordan virker et wattmeter?