


Maskinlæringsmodeller af stof ud over interatomiske potentialer
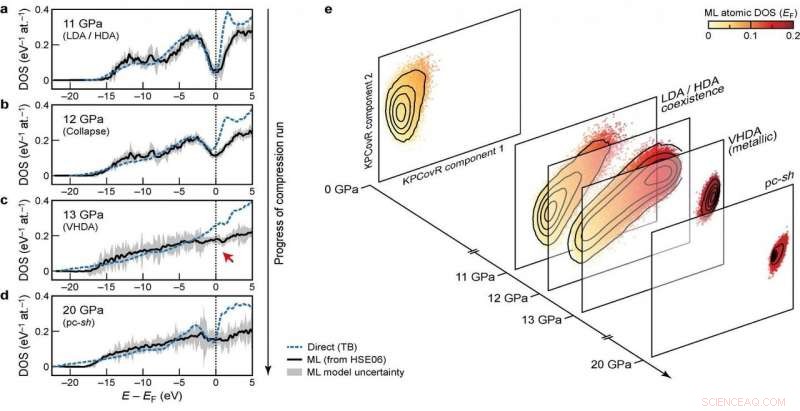
Elektroniske densiteter af tilstande (DOS) på forskellige stadier af kompressionskørslen Kredit:@Michele Ceriotti
Kombination af elektroniske strukturberegninger og machine learning (ML) teknikker er blevet en almindelig tilgang i atomistisk modellering af stof. Ved at bruge de to teknikker sammen har forskere, for eksempel, at skabe modeller, der bruger atomare koordinater som de eneste input til billigt at forudsige enhver egenskab, der kan beregnes ved de første-principper-beregninger, der var blevet brugt til at træne dem.
Mens de tidligste og efterhånden mest avancerede bestræbelser har fokuseret på at bruge forudsigelser af totale energier og atomkræfter til at konstruere interatomiske potentialer, nyere indsats har rettet sig mod yderligere egenskaber ved krystaller og molekyler, såsom ioniseringsenergier, NMR kemiske afskærmninger, dielektriske responsegenskaber og ladningstæthed. I papiret "At lære den elektroniske tæthed af tilstande i kondenseret stof, "Ceriotti og kolleger fokuserer på den elektroniske tæthed af stater (DOS), en anden mængde, der ligger til grund for mange nyttige materialeegenskaber, hvoraf nogle kan observeres eksperimentelt.
DOS er i det væsentlige antallet af forskellige tilstande, som elektroner kan optage på et bestemt energiniveau, og kan bruges, for eksempel, at beregne det elektroniske bidrag til varmekapaciteten i metaller og tætheden af frie ladningsbærere i halvledere. Det er en indirekte proxy for egenskaber såsom energibåndgabet, båndenergien og det optiske absorptionsspektrum.
"Forudsigelse af DOS er en interessant øvelse i sig selv, fordi det i det væsentlige er den enklest mulige beskrivelse af den elektroniske struktur ud over billedtilstandsbilledet, " sagde Ceriotti. "Det er også nyttigt, fordi der er mange egenskaber, som du kan beregne fra DOS, gør det til et godt eksempel på, hvordan den næste generation af ML -modeller kan bruges på samme måde som elektroniske strukturberegninger, bruger dem på en indirekte måde til at beregne mellemliggende mængder, som derefter nemt kan behandles for at evaluere egenskaber, der er sværere at lære direkte."
Ved udviklingen af modellen, gruppen søgte at sikre overførsel på tværs af forskellige faser samt skalerbarhed til store systemstørrelser. Deres ultimative tilgang, som ser på, hvordan forskellige atomkonfigurationer påvirker fordelingen af energiniveauer, opfylder disse mål - det var i stand til at lære og forudsige DFT-beregnet DOS for et forskelligartet datasæt af siliciumstrukturer, dækker en bred vifte af termodynamiske forhold og forskellige faser. Den skalerer også lineært, snarere end med terningen af antallet af atomer som ved elektroniske strukturberegninger, gør den anvendelig til store strukturer. Endelig, modellen tillod en analyse af den lokale DOS, giver forskere mulighed for at undersøge samspillet mellem strukturelle motiver og elektronisk struktur.
Kombinationen af overførbarhed, og skalerbarhed af forudsigelser til store systemstørrelser, gøre modellen anvendelig til at løse mangeårige åbne spørgsmål inden for materialevidenskab. Den nye ramme er allerede blevet brugt til at belyse de elektroniske egenskaber af en 100.000-atoms simulering af amorft silicium, undergår en række faseovergange, når de komprimeres til 20 Gpa, i et papir udgivet i Natur i dag i samarbejde med et team bestående af forskere fra Oxford, Cambridge, US Naval Research Laboratory og Ohio University. Den forudsagte DOS bruges også til at forklare, hvordan de trykinducerede strukturelle transformationer kobles til materialets elektroniske struktur.
Kombinationen af den nye model med en af de veletablerede potentielle energimodeller gør det også muligt at beregne de elektroniske bidrag til makroskopiske egenskaber såsom metallers varmekapacitet og at udføre simuleringer, der tager højde for endelige-elektroniske temperatureffekter – som vist. i en anden snart offentliggjort artikel, der diskuterer nikkels højtemperaturegenskaber. Ja, den nye model er et kritisk skridt i retning af MARVELs mål om at udvikle integrerede maskinlæringsmodeller, der supplerer – og måske i sidste ende erstatter – kostbare elektroniske strukturberegninger.
"Der er andre egenskaber bortset fra elektrontætheden af tilstande, såsom optiske excitationer, og NMR -respons, som vi har været i stand til præcist at forudsige med maskinindlæring. "Ceriotti sagde." Hvis vi kan bruge dem alle i kombination med billige og præcise interatomiske potentialer, vil det give os mulighed for at beskrive alle egenskaber ved materialer med samme nøjagtighed opnået med elektronisk strukturberegning, men til en lille brøkdel af prisen."
 Varme artikler
Varme artikler
-
 Nye egenskaber ved kosmiske stråler, silicium, magnesium og neon fundet af Alpha Magnetic Spectrome…Alpha Magnetic Spectrometer. Kredit:NASA Et meget stort team af forskere fra hele verden har fundet nye egenskaber ved de kosmiske stråler silicium, magnesium og neon ved hjælp af data fra Alpha M
Nye egenskaber ved kosmiske stråler, silicium, magnesium og neon fundet af Alpha Magnetic Spectrome…Alpha Magnetic Spectrometer. Kredit:NASA Et meget stort team af forskere fra hele verden har fundet nye egenskaber ved de kosmiske stråler silicium, magnesium og neon ved hjælp af data fra Alpha M -
 Forskere gennemfører første fase af eksperimentet for at studere kæmpe luftbrusereKredit:National Research Nuclear University Den første fase af en undersøgelse af kæmpe luftbrusere er afsluttet på NEVOD-SHAL, en ny facilitet oprettet på det nationale forskningsnukleare univers
Forskere gennemfører første fase af eksperimentet for at studere kæmpe luftbrusereKredit:National Research Nuclear University Den første fase af en undersøgelse af kæmpe luftbrusere er afsluttet på NEVOD-SHAL, en ny facilitet oprettet på det nationale forskningsnukleare univers -
 Ny forskning tilføjer Prandtls arbejde, far til moderne aerodynamikKonturvisualisering af blandet tilstand ustabilitet i Prandtl-modellen for en hældningsvinkel på 30 (grader). Flow er fra top til bund. Vortikale strukturer identificeres ved hjælp af Q-kriteriet. Kre
Ny forskning tilføjer Prandtls arbejde, far til moderne aerodynamikKonturvisualisering af blandet tilstand ustabilitet i Prandtl-modellen for en hældningsvinkel på 30 (grader). Flow er fra top til bund. Vortikale strukturer identificeres ved hjælp af Q-kriteriet. Kre -
 Forsinket tilpasning fremmer sameksistensKredit:Mick Lissone/public domain Jordbakterier skal kunne tilpasse sig varierende miljøforhold. - Men en ny undersøgelse fra LMU-forskere indikerer, at hurtig tilpasning kan være kontraproduktiv,
Forsinket tilpasning fremmer sameksistensKredit:Mick Lissone/public domain Jordbakterier skal kunne tilpasse sig varierende miljøforhold. - Men en ny undersøgelse fra LMU-forskere indikerer, at hurtig tilpasning kan være kontraproduktiv,