


Bondings næste topmodel:Projektering af bindingsegenskaber med maskinlæring

Forskere fra University of Tokyo Institute of Industrial Science rapporterer om en maskinlæringsbaseret model til at forudsige materialers bindingsegenskaber. Kredit:Institut for Industrividenskab, universitetet i Tokyo
At designe materialer, der har de nødvendige egenskaber til at opfylde specifikke funktioner, er en udfordring for forskere, der arbejder inden for områder fra katalyse til solceller. For at fremskynde udviklingsprocesser, modelleringstilgange kan bruges til at forudsige information for at vejlede forbedringer. Forskere fra University of Tokyo Institute of Industrial Science har udviklet en maskinlæringsmodel til at bestemme karakteristika for bundne og adsorberede materialer baseret på parametre for de enkelte komponenter. Deres resultater er offentliggjort i Anvendt Fysik Express .
Faktorer som længden og styrken af bindinger i materialer spiller afgørende roller for at bestemme de strukturer og egenskaber, vi oplever på den makroskopiske skala. Evnen til nemt at forudsige disse egenskaber er derfor værdifuld, når man designer nye materialer.
Densiteten af tilstande (DOS) er en parameter, der kan beregnes for individuelle atomer, molekyler, og materialer. Enkelt sagt, den beskriver de muligheder, der er til rådighed for elektronerne, der arrangerer sig i et materiale. En modelleringstilgang, der kan tage denne information for udvalgte komponenter og producere nyttige data for det ønskede produkt - uden behov for at fremstille og analysere materialet - er et attraktivt værktøj.
Forskerne brugte en maskinlæringstilgang – hvor modellen forfiner sin respons uden menneskelig indgriben – til at forudsige fire forskellige egenskaber ved produkter ud fra de individuelle komponenters DOS-information. Selvom DOS er blevet brugt som en deskriptor til at etablere enkelte parametre før, det er første gang, at flere forskellige egenskaber er blevet forudsagt.
"Vi var i stand til kvantitativt at forudsige bindingsenergien, bindingslængde, antal kovalente elektroner, og Fermi-energien efter binding for tre forskellige generelle systemtyper, " forklarer undersøgelsens første forfatter Eiki Suzuki. "Og vores forudsigelser var meget nøjagtige på tværs af alle ejendommene."
Fordi beregningen af DOS i en isoleret tilstand er mindre kompleks end for bundne systemer, analysen er relativt effektiv. Ud over, den anvendte neurale netværksmodel fungerede godt, selv når kun 20 % af datasættet blev brugt til træning.
"En væsentlig fordel ved vores model er, at den er generel og kan anvendes på en lang række systemer, ", forklarer den tilsvarende forfatter Teruyasu Mizoguchi. "Vi mener, at vores resultater kan yde et væsentligt bidrag til adskillige udviklingsprocesser, for eksempel i katalyse, og kunne være særlig nyttig i nyere forskningsområder som nano-klynger og nanotråde."
Artiklen, "Nøjagtig forudsigelse af bindingsegenskaber ved hjælp af en maskinlæringsbaseret model ved brug af isolerede tilstande før binding", blev udgivet i Anvendt Fysik Express .
 Varme artikler
Varme artikler
-
 Udstrakte fotoner genopretter tabt interferensForskere registrerede disse mønstre af kvanteinterferens mellem tre fotoner, der startede som separate, skelnelige partikler. Kredit:Joint Quantum Institute De mindste stykker af naturen - individ
Udstrakte fotoner genopretter tabt interferensForskere registrerede disse mønstre af kvanteinterferens mellem tre fotoner, der startede som separate, skelnelige partikler. Kredit:Joint Quantum Institute De mindste stykker af naturen - individ -
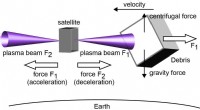 Plasma thruster:Ny teknologi til fjernelse af rumaffaldEt koncept til fjernelse af rumaffald ved tovejs impulsudslyngning fra en satellit. Kredit:Kazunori Takahashi. Jorden er i øjeblikket omgivet af affald, der er sendt ud i rummet gennem flere årtie
Plasma thruster:Ny teknologi til fjernelse af rumaffaldEt koncept til fjernelse af rumaffald ved tovejs impulsudslyngning fra en satellit. Kredit:Kazunori Takahashi. Jorden er i øjeblikket omgivet af affald, der er sendt ud i rummet gennem flere årtie -
 Maskinlæring for at opskalere kvantecomputeren, , Et kort over elektronbølgefunktionsmønstre, hvor symmetrien, lysstyrke og størrelse af funktioner er direkte relateret til positionen af et fosforatom i siliciumgitteret. Kredit:M.Usman/ Univ
Maskinlæring for at opskalere kvantecomputeren, , Et kort over elektronbølgefunktionsmønstre, hvor symmetrien, lysstyrke og størrelse af funktioner er direkte relateret til positionen af et fosforatom i siliciumgitteret. Kredit:M.Usman/ Univ -
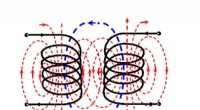 Elektromagnetisk trolddom:Trådløs strømoverførsel forbedret af bagudgående signalStiplede linjer af magnetfelterne omkring to induktionsspoler illustrerer princippet om elektromagnetisk induktion. Kredit:Alex Krasnok et al./ Fysisk gennemgangsbreve Et internationalt forskerh
Elektromagnetisk trolddom:Trådløs strømoverførsel forbedret af bagudgående signalStiplede linjer af magnetfelterne omkring to induktionsspoler illustrerer princippet om elektromagnetisk induktion. Kredit:Alex Krasnok et al./ Fysisk gennemgangsbreve Et internationalt forskerh