


Ny kvantealgoritme løser kritiske kvantekemiproblemer gennem tilpasning langs en geometrisk vej
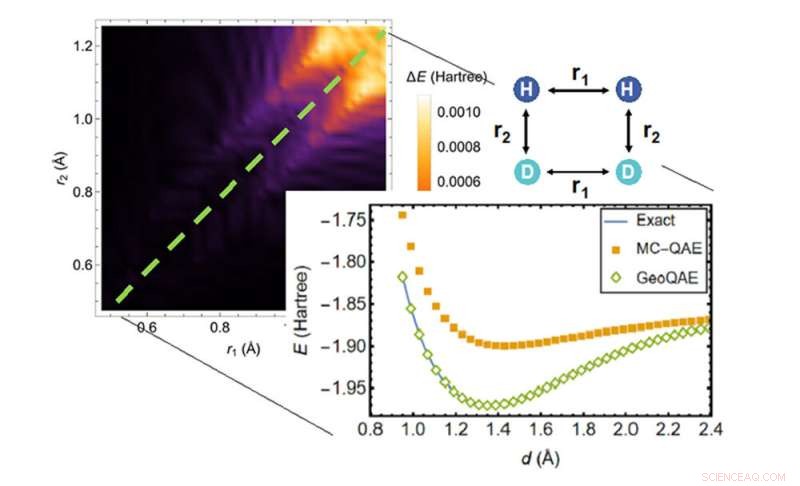
Ved beregning af den potentielle energioverflade af den kemiske reaktion af H2;+ D2 → 2HD udkonkurrerer den nye algoritme (grønne diamanter) den tidligere algoritme (orange firkanter) ved at finde den mest nøjagtige løsning (blå linje). Kredit:Brookhaven National Laboratory
Et team af forskere fra det amerikanske energiministeriums (DOE) Brookhaven National Laboratory og Stony Brook University har udtænkt en ny kvantealgoritme til at beregne de laveste energier af molekyler ved specifikke konfigurationer under kemiske reaktioner, herunder når deres kemiske bindinger brydes. Som beskrevet i Physical Review Research , sammenlignet med lignende eksisterende algoritmer, inklusive holdets tidligere metode, vil den nye algoritme markant forbedre videnskabsmænds evne til nøjagtigt og pålideligt at beregne den potentielle energioverflade i reagerende molekyler.
Til dette arbejde arbejdede Deyu Lu, en Center for Functional Nanomaterials (CFN) fysiker ved Brookhaven Lab, sammen med Tzu-Chieh Wei, en lektor med speciale i kvanteinformationsvidenskab ved C.N. Yang Institut for Teoretisk Fysik ved Stony Brook University, Qin Wu, en teoretiker ved CFN, og Hongye Yu, en Ph.D. studerende ved Stony Brook.
"Forståelse af kvantemekanikken i et molekyle, hvordan det opfører sig på atomniveau, kan give nøgleindsigt i dets kemiske egenskaber, såsom dets stabilitet og reaktivitet," sagde Lu.
En særlig egenskab, der har været en udfordring at bestemme, er et molekyles grundtilstand:det punkt, hvor molekylets samlede elektroniske energi (inklusive kinetisk og potentiel energi) er på sit laveste, og intet uden for det "molekylære system" er spændende eller oplader molekylets elektroner. Når atomstrukturen i et kemisk system bliver mere kompleks, som i et stort molekyle, kan mange flere elektroner interagere. Disse interaktioner gør det ekstremt vanskeligt at beregne grundtilstanden for komplekse molekyler.
Den nye kvantealgoritme forbedrer den tidligere algoritme for at tackle dette problem på en kreativ måde. Det udnytter en jævn, geometrisk deformation lavet af kontinuerligt varierende bindingslængder eller bindingsvinkler i molekylets struktur. Med denne tilgang siger forskerne, at de kan beregne grundtilstanden for molekyler meget nøjagtigt, selvom kemiske bindinger brydes og omdannes under kemiske reaktioner.
Bygge grundlaget
"Når man udelukkende stoler på traditionelle computermetoder, indeholder dette grundtilstandsproblem for mange variabler til at løse - selv på de mest kraftfulde supercomputere," sagde Lu.
Du kan tænke på en algoritme som et sæt trin til at løse et bestemt problem. Klassiske computere kan køre komplekse algoritmer, men efterhånden som de bliver større og mere involverede, kan de blive for svære eller tidskrævende for klassiske computere at løse. Kvantecomputere kan fremskynde processen ved at udnytte kvantemekanikkens regler.
I klassisk databehandling lagres data i bits, der har en værdi på 1 eller 0. En kvantebit, kendt som en qubit, kan have en værdi ud over blot 0 eller 1, den kan endda have en værdi på 0 og 1, i en såkaldt kvantesuperposition. I princippet kan disse mere "fleksible" qubits lagre en større mængde information end klassiske bits. Hvis videnskabsmænd kan finde måder at udnytte qubits informationsbærende kapacitet, kan computerkraft udvides eksponentielt med hver yderligere qubit.
Qubits er dog ret skrøbelige. De kan ofte gå i stykker, når information bliver udtrukket. Når en kvanteenhed interagerer med det omgivende miljø, kan den generere støj eller interferens, der ødelægger kvantetilstanden. Temperaturændringer, vibrationer, elektromagnetisk interferens og endda materialefejl kan også få qubits til at miste information.
For at kompensere for disse faldgruber udviklede forskere en hybridløsning, der udnytter både klassiske computeralgoritmer, som er mere stabile og praktiske.
Lu og Wei begyndte at forske i hybride klassiske og kvanteberegningsmetoder i 2019. Denne årlige bevilling fremmer samarbejdet mellem Brookhaven National Laboratory og Stony Brook University ved at finansiere fælles forskningsinitiativer, der stemmer overens med begge institutioners missioner. Med dette indledende arbejde fokuserede Lu og Wei først på at løse grundtilstandsproblemet ved at erstatte de mest "dyre" klassiske algoritmer - dem, der var meget mere komplekse og krævede betydeligt flere trin (og mere computertid) at gennemføre - med kvantealgoritmer .
Strækker bånd, skaber nye veje
Forskerne bemærker, at eksisterende kvantealgoritmer alle har ulemper for at løse grundtilstandsproblemet, inklusive den, Wei og Yu udviklede i 2019. Mens nogle populære algoritmer er nøjagtige, når et molekyle er i sin ligevægtsgeometri - dets naturlige arrangement af atomer i tre dimensioner - disse algoritmer kan blive upålidelige, når de kemiske bindinger brydes ved store atomare afstande. Bindingsdannelse og dissociation spiller en rolle i mange applikationer, såsom at forudsige, hvor meget energi det tager at få en kemisk reaktion i gang, så forskerne havde brug for en måde at tackle dette problem på, når molekyler reagerer. De havde brug for nye kvantealgoritmer, der kan beskrive bindingsbrud.
Til denne nye version af algoritmen arbejdede teamet med det Brookhaven-Lab-ledede Co-design Center for Quantum Advantage (C2QA), som blev dannet i 2020. Wei bidrager til centrets software-thrust, som er specialiseret i kvantealgoritmer. Holdets nye algoritme bruger en adiabatisk tilgang – en der foretager gradvise ændringer – men med nogle tilpasninger, der sikrer, at den forbliver pålidelig, når kemiske bindinger brydes.
"En adiabatisk proces fungerer ved gradvist at tilpasse betingelserne for et kvantemekanisk system," forklarede Lu. "På en måde når man en løsning i meget små trin. Man udvikler systemet fra en simpel, løsbar model til det endelige mål, typisk en sværere model. Udover grundtilstanden dog et mange-elektronisk system har mange exciterede tilstande ved højere energier. Disse exciterede tilstande kan udgøre en udfordring, når du bruger denne metode til at beregne grundtilstanden."
Wei sammenlignede en adiabatisk algoritme med at køre langs en motorvej, "hvis du rejser fra den ene by til den næste, er der flere veje til at komme dertil, men du vil gerne finde den sikreste og mest effektive."
I tilfælde af kvantekemi er nøglen at finde et stort nok "energigab" mellem grundtilstanden og exciterede tilstande, hvor der ikke eksisterer nogen elektrontilstande. Med et stort nok mellemrum vil køretøjerne i motorvejsmetaforen ikke "krydse vejbaner", så deres stier kan spores nøjagtigt.
"Et stort mellemrum betyder, at du kan køre hurtigere, så på en måde forsøger du at finde en mindre befærdet motorvej for at køre hurtigere uden at ramme noget," sagde Wei.
"Med disse algoritmer er indgangen til stien en veldefineret, enkel løsning fra klassisk databehandling," bemærkede Wei. "Vi ved også, hvor udgangen er - molekylets grundtilstand - og vi forsøgte at finde en måde at forbinde det med indgangen på den mest naturlige måde, en lige linje.
"Det gjorde vi i vores første papir, men den lige linje havde vejspærringer forårsaget af, at energigabet blev lukket og stier krydsede. Nu har vi en bedre løsning."
Da forskerne testede algoritmen, viste de, at selv med endelige bindingslængdeændringer fungerede den forbedrede version stadig nøjagtigt for grundtilstanden.
"Vi gik ud over vores komfortzone, fordi kemi ikke er vores fokus," sagde Wei. "Men det var godt at finde en ansøgning som denne og fremme denne form for samarbejde med CFN. Det er vigtigt at have forskellige perspektiver i forskningen."
Han bemærkede den akkumulerede indsats fra mange mennesker. "I den store ordning tror jeg, at vi yder et lille bidrag, men dette kunne være et grundlag for andet arbejde på disse områder," sagde han. "Denne forskning er ikke kun grundlæggende, men en fantastisk illustration af, hvordan forskellige institutioner og faciliteter kan mødes for at udnytte deres ekspertiseområder." + Udforsk yderligere
Mod en kvantecomputer, der beregner molekylær energi
Sidste artikelBygger bedre kvantesensorer
Næste artikelEn ny eksperimentel undersøgelse tackler nanoboblernes uløste mysterium
 Varme artikler
Varme artikler
-
 Tuning terahertz transmission(venstre) En monteret enhed inklusive den nye justerbare metasurface udviklet af Ding, Teng og kolleger. (højre) Når terahertz-stråling rammer overfladen af indbyrdes forbundne p-type og n-type halv
Tuning terahertz transmission(venstre) En monteret enhed inklusive den nye justerbare metasurface udviklet af Ding, Teng og kolleger. (højre) Når terahertz-stråling rammer overfladen af indbyrdes forbundne p-type og n-type halv -
 Forstå måden, hvorpå væske spredes gennem papirEfterforskere dokumenterede spredning af blæk på et filterpapir af Whatman Grade 1, og analyserede processen med et scanningselektronmikroskop. Scanning af mikrografiske resultater viser distribution
Forstå måden, hvorpå væske spredes gennem papirEfterforskere dokumenterede spredning af blæk på et filterpapir af Whatman Grade 1, og analyserede processen med et scanningselektronmikroskop. Scanning af mikrografiske resultater viser distribution -
 Produktion af radioisotoper til medicinsk billeddannelse og sygdomsbehandlingCathy Cutler, Lisa Muench, Tatjana Klaric, Weimin Zhou, Vicky Litton, og Anna Goldberg i hot cell -området, hvor BLIP -mål behandles for at udtrække ønskede isotopprodukter. Kredit:Brookhaven National
Produktion af radioisotoper til medicinsk billeddannelse og sygdomsbehandlingCathy Cutler, Lisa Muench, Tatjana Klaric, Weimin Zhou, Vicky Litton, og Anna Goldberg i hot cell -området, hvor BLIP -mål behandles for at udtrække ønskede isotopprodukter. Kredit:Brookhaven National -
 Forskere observerer interferenseffekt mellem Floquet kvasipartikler ved hjælp af strontium optisk g…Skematisk billede af eksperimentopsætning og interferens. Kredit:NTSC Baseret på strontium optisk gitter ur platform, et forskerhold ledet af prof. Chang Hong fra National Time Service Center for
Forskere observerer interferenseffekt mellem Floquet kvasipartikler ved hjælp af strontium optisk g…Skematisk billede af eksperimentopsætning og interferens. Kredit:NTSC Baseret på strontium optisk gitter ur platform, et forskerhold ledet af prof. Chang Hong fra National Time Service Center for
- Find galakser med aktive kerner
- Fysikere tilbyder et nyt spin på hukommelsen
- Ny måde at fremstille biomedicinsk udstyr på af silke giver bedre produkter med afstembare kvalite…
- Brunst minedæmning i Sydafrika:Hvad skal der gøres for at forhindre endnu en katastrofe
- Fem døde efter en sjælden tornado ramte Tjekkiet
- Med ny optisk enhed, ingeniører kan finjustere lysets farve