


Forudsigelse af sekvens fra struktur
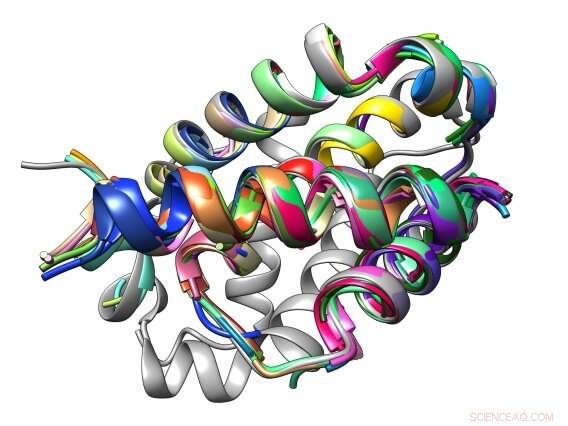
Bindingsgrænsefladen mellem et peptid og dets Bcl-2-proteinmål er sammensat af almindelige strukturelle motiver kendt som TERM'er. Kredit:Sebastian Swanson og Avi Singer
En måde at undersøge indviklede biologiske systemer på er at blokere deres komponenter i at interagere og se, hvad der sker. Denne metode giver forskere mulighed for bedre at forstå cellulære processer og funktioner, udvidelse af daglige laboratorieeksperimenter, diagnostiske analyser, og terapeutiske interventioner. Som resultat, reagenser, der hindrer interaktioner mellem proteiner, er i høj efterspørgsel. Men før videnskabsmænd hurtigt kan generere deres egne tilpassede molekyler, der er i stand til at gøre det, de skal først analysere det komplicerede forhold mellem sekvens og struktur.
Små molekyler kan nemt trænge ind i celler, men grænsefladen, hvor to proteiner binder til hinanden, er ofte for stor eller mangler de små hulrum, der kræves for, at disse molekyler kan målrettes. Antistoffer og nanostoffer binder sig til længere strækninger af protein, hvilket gør dem bedre egnede til at hindre protein-protein-interaktioner, men deres store størrelse og komplekse struktur gør dem vanskelige at levere og ustabile i cytoplasmaet. Derimod korte strækninger af aminosyrer, kendt som peptider, er store nok til at binde lange strækninger af protein, mens de stadig er små nok til at komme ind i celler.
Keating-laboratoriet på MIT Institut for Biologi arbejder hårdt på at udvikle måder til hurtigt at designe peptider, der kan forstyrre protein-protein-interaktioner, der involverer Bcl-2-proteiner, som fremmer kræftvækst. Deres seneste tilgang bruger et computerprogram kaldet dTERMen, udviklet af Keating lab alumnus, Gevorg Grigoryan Ph.D. '07, i øjeblikket lektor i datalogi og adjungeret lektor i biologiske videnskaber og kemi ved Dartmouth College. Forskere giver simpelthen programmet deres ønskede strukturer, og det spytter aminosyresekvenser ud for peptider, der er i stand til at forstyrre specifikke protein-protein-interaktioner.
"Det er sådan en enkel tilgang at bruge, " siger Keating, en MIT-professor i biologi og seniorforfatter på undersøgelsen. "I teorien, du kan indsætte en hvilken som helst struktur og løse en sekvens. I vores undersøgelse, programmet kom med nye sekvenskombinationer, der ikke ligner noget, der findes i naturen – det udledte en helt unik måde at løse problemet på. Det er spændende at afsløre nye territorier i sekvensuniverset."
Tidligere postdoc Vincent Frappier og Justin Jenson Ph.D. '18 er med-første forfattere på undersøgelsen, som udkommer i det seneste nummer af Struktur .
Samme problem, anderledes tilgang
Jenson, for hans del, har tacklet udfordringen med at designe peptider, der binder til Bcl-2-proteiner ved hjælp af tre forskellige tilgange. Den dTERMen-baserede metode, han siger, er langt den mest effektive og generelle, han har prøvet endnu.
Standardtilgange til at opdage peptidhæmmere involverer ofte modellering af hele molekyler ned til fysikken og kemien bag individuelle atomer og deres kræfter. Andre metoder kræver tidskrævende screeninger for de bedste bindingskandidater. I begge tilfælde processen er besværlig, og succesraten er lav.
dTERMænd, derimod kræver hverken fysik eller eksperimentel screening, og udnytter fælles enheder af kendte proteinstrukturer, som alfa-helixer og beta-strenge - kaldet tertiære strukturelle motiver eller "TERMs" - som er samlet i samlinger som Protein Data Bank. dTERMen udtrækker disse strukturelle elementer fra databanken og bruger dem til at beregne, hvilke aminosyresekvenser der kan antage en struktur, der er i stand til at binde sig til og afbryde specifikke protein-protein-interaktioner. Det tager en enkelt dag at bygge modellen, og få sekunder til at evaluere tusind sekvenser eller designe et nyt peptid.
"dTERMen giver os mulighed for at finde sekvenser, der sandsynligvis har de bindingsegenskaber, vi leder efter, i en robust, effektiv, og generel måde med høj succesrate, " siger Jenson. "Tidligere tilgange har taget år. Men ved at bruge dTERMen, vi gik fra strukturer til validerede designs i løbet af få uger."
Af de 17 peptider de byggede ved hjælp af de designede sekvenser, 15 bundet med indfødt-lignende affinitet, forstyrre Bcl-2 protein-protein-interaktioner, som er notorisk svære at målrette mod. I nogle tilfælde, deres designs var overraskende selektive og bundet til et enkelt Bcl-2 familiemedlem frem for de andre. De designede sekvenser afveg fra kendte sekvenser fundet i naturen, hvilket i høj grad øger antallet af mulige peptider.
"Denne metode tillader en vis grad af fleksibilitet, " siger Frappier. "dTERMen er mere robuste over for strukturelle ændringer, som giver os mulighed for at udforske nye typer strukturer og diversificere vores portefølje af potentielle bindende kandidater."
Undersøgelse af sekvensuniverset
I betragtning af de terapeutiske fordele ved at hæmme Bcl-2-funktionen og bremse tumorvækst, Keating-laboratoriet er allerede begyndt at udvide deres designberegninger til andre medlemmer af Bcl-2-familien. De agter på sigt at udvikle nye proteiner, der vedtager strukturer, som aldrig er set før.
"Vi har nu set nok eksempler på forskellige lokale proteinstrukturer til, at beregningsmodeller af sekvens-strukturforhold kan udledes direkte fra strukturelle data, snarere end at skulle genopdages hver gang fra atomistiske interaktionsprincipper, " siger Grigoryan, dTERMen's skaber. "Det er uhyre spændende, at en sådan strukturbaseret slutning virker og er nøjagtig nok til at muliggøre robust proteindesign. Det giver et fundamentalt anderledes værktøj til at hjælpe med at tackle strukturbiologiens nøgleproblemer - fra proteindesign til strukturforudsigelse."
Frappier håber en dag at være i stand til at screene hele det menneskelige proteom beregningsmæssigt, ved at bruge metoder som dTERMen til at generere kandidatbindingspeptider. Jenson foreslår, at brug af dTERMen i kombination med mere traditionelle tilgange til sekvens-redesign kunne forstærke et allerede kraftfuldt værktøj, bemyndigelse af forskere til at producere disse målrettede peptider. Ideelt set han siger, en dag at udvikle peptider, der binder og hæmmer dit yndlingsprotein, kunne være lige så nemt som at køre et computerprogram, eller lige så rutinepræget som at designe en DNA-primer.
Ifølge Keating, selvom den tid stadig ligger i fremtiden, "vores undersøgelse er det første skridt mod at demonstrere denne kapacitet på et problem af beskedent omfang."
Denne historie er genudgivet med tilladelse fra MIT News (web.mit.edu/newsoffice/), et populært websted, der dækker nyheder om MIT-forskning, innovation og undervisning.
Sidste artikelGenetik indsats beriger ernæring af popcorn, sorghum
Næste artikelNy enhed forenkler måling af fluorforurening i vand
 Varme artikler
Varme artikler
-
 Forskere gør plastflaskeaffald til ultralet supermateriale med vidtrækkende applikationerEt team ledet af forskere fra National University of Singapore har fundet en måde at forvandle plastflaskeaffald til ultralette polyethylenterephthalat (PET) aerogeler, der er velegnede til forskellig
Forskere gør plastflaskeaffald til ultralet supermateriale med vidtrækkende applikationerEt team ledet af forskere fra National University of Singapore har fundet en måde at forvandle plastflaskeaffald til ultralette polyethylenterephthalat (PET) aerogeler, der er velegnede til forskellig -
 Forståelse af vands mærkelige adfærdEn clathrate is, med oxygener repræsenteret som kugler, og hydrogenbindinger som linier. Arbejdet har vist, hvordan komplekse krystallinske strukturer opstår som følge af vands interaktioner. Kredit:U
Forståelse af vands mærkelige adfærdEn clathrate is, med oxygener repræsenteret som kugler, og hydrogenbindinger som linier. Arbejdet har vist, hvordan komplekse krystallinske strukturer opstår som følge af vands interaktioner. Kredit:U -
 Mekanisme bag platinkatalysator fangetTil venstre:Billede lavet med et Scanning Tunneling Microscope (STM). Billede af en platinoverflade under et tryk på 1 iltatmosfære ved 256 ° C. Under disse omstændigheder, vi ser spontan vækst af en
Mekanisme bag platinkatalysator fangetTil venstre:Billede lavet med et Scanning Tunneling Microscope (STM). Billede af en platinoverflade under et tryk på 1 iltatmosfære ved 256 ° C. Under disse omstændigheder, vi ser spontan vækst af en -
 Nye kuldioxid-adsorberende krystaller til biomedicinske materialer, der er afhængige af formhukomme…Forskere dokumenterede, hvordan et porøst materiale kan ændre sig og bevare sin form, selv efter at have absorberet og frigivet kuldioxid. Her, krystallens porer forbliver åbne efter at have frigivet
Nye kuldioxid-adsorberende krystaller til biomedicinske materialer, der er afhængige af formhukomme…Forskere dokumenterede, hvordan et porøst materiale kan ændre sig og bevare sin form, selv efter at have absorberet og frigivet kuldioxid. Her, krystallens porer forbliver åbne efter at have frigivet
- Nu er et smart pæresystem blevet hacket
- Perfekt dopede kvanteprikker giver farver at farve til
- Klima, græsser og tænder - udviklingen af pattedyr i Sydamerika
- Få dit lægemiddel nano-leveret via mikroboble
- Hvorfor er det vigtigt at kalibrere en pH-meter og dens elektroder mod en buffer?
- Undersøgelse udforsker regnbuebølger og identitetsgab i LGBTQ-liberale politiske perspektiver