


Maskinlæring fremskynder simuleringer inden for materialevidenskab
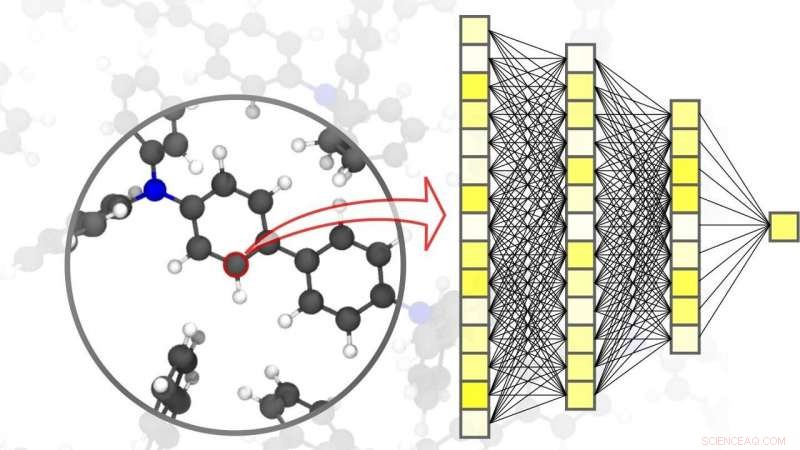
Neurale netværk muliggør præcise simuleringer inden for materialevidenskab – ned til niveauet af individuelle atomer. Kredit:Pascal Friedrich, SÆT
Forskning, udvikling, og produktion af nye materialer afhænger i høj grad af tilgængeligheden af hurtige og samtidig nøjagtige simuleringsmetoder. Maskinelæring, hvor kunstig intelligens (AI) selvstændigt erhverver og anvender ny viden, vil snart sætte forskere i stand til at udvikle komplekse materialesystemer i et rent virtuelt miljø. Hvordan virker det, og hvilke applikationer vil gavne? I en artikel offentliggjort i Naturmaterialer tidsskrift, en forsker fra Karlsruhe Institute of Technology (KIT) og hans kolleger fra Göttingen og Toronto forklarer det hele.
Digitalisering og virtualisering bliver stadig vigtigere i en lang række videnskabelige discipliner. En af disse discipliner er materialevidenskab:forskning, udvikling, og produktion af nye materialer afhænger i høj grad af tilgængeligheden af hurtige og samtidig nøjagtige simuleringsmetoder. Det her, på tur, er gavnlig til en lang række forskellige anvendelser – lige fra effektive energilagringssystemer, såsom dem, der er uundværlige for brugen af vedvarende energi, til ny medicin, for hvis udvikling en forståelse af komplekse biologiske processer er påkrævet. AI og maskinlæringsmetoder kan tage simuleringer i materialevidenskab til det næste niveau. "Sammenlignet med konventionelle simuleringsmetoder baseret på klassiske eller kvantemekaniske beregninger, brugen af neurale netværk specielt skræddersyet til materialesimuleringer gør det muligt for os at opnå en betydelig hastighedsfordel, " forklarer fysiker og AI-ekspert professor Pascal Friederich, Leder af forskningsgruppen AiMat—Artificial Intelligence for Materials Sciences ved KIT's Institute of Theoretical Informatics (ITI). "Med hurtigere simuleringssystemer, forskere vil være i stand til at udvikle større og mere komplekse materialesystemer i et rent virtuelt miljø, og at forstå og optimere dem ned til det atomare niveau."
Høj præcision fra atomet til materialet
I en artikel offentliggjort i Naturmaterialer , Pascal Friedrich, som også er associeret gruppeleder for Nanomaterials by Information-Guided Design-afdelingen ved KIT's Institute of Nanotechnology (INT), gaver, sammen med forskere fra University of Göttingen og University of Toronto, en oversigt over de grundlæggende principper for maskinlæring brugt til simuleringer i materialevidenskab. Dette omfatter også dataopsamlingsprocessen og aktive læringsmetoder. Maskinlæringsalgoritmer gør det ikke kun muligt for kunstig intelligens at behandle inputdata, men også for at finde mønstre og sammenhænge i store datasæt, lære af dem, og foretage selvstændige forudsigelser og beslutninger. For simuleringer i materialevidenskab, det er vigtigt at opnå høj nøjagtighed over forskellige tids- og størrelsesskalaer, lige fra atomet til materialet, mens beregningsomkostningerne begrænses. I deres artikel, forskerne diskuterer også forskellige aktuelle anvendelser, såsom små organiske molekyler og store biomolekyler, strukturelt forstyrret fast stof, væske, og gasformige materialer, såvel som komplekse krystallinske systemer – f.eks. metalorganiske rammer, der kan bruges til gaslagring eller til separation, til sensorer eller til katalysatorer.
Endnu mere hastighed med hybridmetoder
For yderligere at udvide mulighederne for materialesimuleringer i fremtiden, forskerne fra Karlsruhe, Göttingen, og Toronto foreslår udviklingen af hybride metoder:disse kombinerer maskinlæring (ML) og molekylær mekanik (MM) metoder. MM-simuleringer bruger såkaldte kraftfelter for at beregne de kræfter, der virker på hver enkelt partikel og dermed forudsige bevægelser. Da potentialerne for ML- og MM-metoderne er ret ens, en tæt integration med variable overgangsområder er mulig. Disse hybridmetoder kan markant accelerere simuleringen af store biomolekyler eller enzymatiske reaktioner i fremtiden, for eksempel.
 Varme artikler
Varme artikler
-
 Undersøgelse af porerne i membranvesiklerMikrograf, der viser celler i det murine immunsystem. Molekylet YM201636 forstørrer selektivt de grønne farvede sene endosomer og lysosomer (til højre), mens et specifikt par biotoksiner kun virker på
Undersøgelse af porerne i membranvesiklerMikrograf, der viser celler i det murine immunsystem. Molekylet YM201636 forstørrer selektivt de grønne farvede sene endosomer og lysosomer (til højre), mens et specifikt par biotoksiner kun virker på -
 Power-to-gas anlæg med høj effektivitetDemonstrationsfaciliteten i HELMETH-projektet kombinerer metanering (venstre) og elektrolyse (højre) med en effektivitet på 76 procent. Kredit:sunfire GmbH Naturgasnettet kan fungere som buffer fo
Power-to-gas anlæg med høj effektivitetDemonstrationsfaciliteten i HELMETH-projektet kombinerer metanering (venstre) og elektrolyse (højre) med en effektivitet på 76 procent. Kredit:sunfire GmbH Naturgasnettet kan fungere som buffer fo -
 Drysset med kraft:Hvordan urenheder forstærker et termoelektrisk materiale på atomniveauHAXPES på Spring-8. Kredit:Dr Kotsugi I søgen efter løsninger på stadigt forværrede miljøproblemer, såsom udtømning af fossile brændstoffer og klimaændringer, mange har vendt sig til termoelektris
Drysset med kraft:Hvordan urenheder forstærker et termoelektrisk materiale på atomniveauHAXPES på Spring-8. Kredit:Dr Kotsugi I søgen efter løsninger på stadigt forværrede miljøproblemer, såsom udtømning af fossile brændstoffer og klimaændringer, mange har vendt sig til termoelektris -
 Forskere udvikler ny chip til overlegen retsmedicinsk påvisning af blodresterBGU-mikrofluidchippen øger ikke kun den kemiluminescerende intensitet flere gange, men forlænger også luminols glødetid, muliggør påvisning af meget mindre blodprøver i en retsmedicinsk scene. Chipenh
Forskere udvikler ny chip til overlegen retsmedicinsk påvisning af blodresterBGU-mikrofluidchippen øger ikke kun den kemiluminescerende intensitet flere gange, men forlænger også luminols glødetid, muliggør påvisning af meget mindre blodprøver i en retsmedicinsk scene. Chipenh
- Europas energigiganter høster fordele af olieprisstigninger
- Bakteriefabrikker kunne fremstille højtydende proteiner til rummissioner
- Digitalisering af sociale ydelser kan yderligere udelukke folk, der allerede er på kanten
- Effekten af saltholdighed på Photosynthesis
- Receptpligtig medicin fundet i Yorks floder
- Rumvandrende astronauter opsætter tv-kameraer til ankommende skibe