


Billeddannelse af kemiske fingeraftryk af molekyler
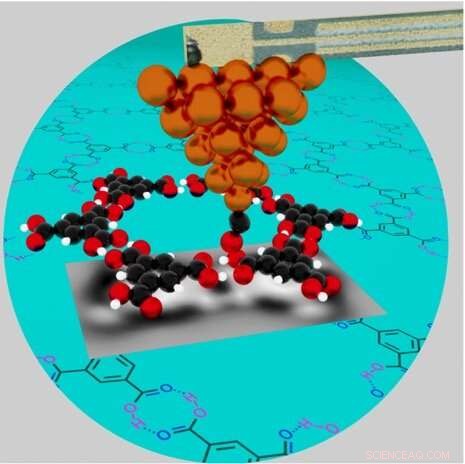
En illustration af et højopløseligt atomkraftmikroskop, der undersøger de kemiske egenskaber af hydrogenbundet trimesinsyre (TMA) netværk (overlejret på blågrøn cirkel) på en kobberoverflade. Nøgle:kobberatomer på metalspidsens top (orange), kulstofatomer (sort), oxygenatomer (røde) og brintatomer (hvide). Det enkelte carbonmonoxid (CO)-molekyle for enden af spidsens spids, med carbonet knyttet til kobber, er en smule bøjet som reaktion på de frastødende kræfter fra TMA-molekylets nærliggende oxygen. Kredit:Brookhaven National Laboratory
Bladr gennem enhver lærebog i kemi, og du vil se tegninger af molekylers kemiske struktur – hvor individuelle atomer er arrangeret i rummet, og hvordan de er kemisk bundet til hinanden. I årtier kunne kemikere kun indirekte bestemme kemiske strukturer baseret på responsen, der blev genereret, når prøver interagerede med røntgenstråler eller partikler af lys. Til det specielle tilfælde af molekyler på en overflade gav atomkraftmikroskopi (AFM), opfundet i 1980'erne, direkte billeder af molekyler og de mønstre, de danner, når de samles i todimensionelle (2D) arrays. I 2009 gjorde betydelige fremskridt inden for højopløsnings-AFM (HR-AFM) det muligt for kemikere for første gang at afbilde den kemiske struktur af et enkelt molekyle direkte med tilstrækkelige detaljer til at skelne mellem forskellige typer binding inde i molekylet.
AFM "føler" kræfterne mellem en skarp sondespids og overfladeatomer eller molekyler. Spidsen scanner over en prøveoverflade, venstre mod højre og top til bund, i en højde på mindre end en nanometer, og registrerer kraften i hver position. En computer kombinerer disse målinger for at generere et kraftkort, hvilket resulterer i et øjebliksbillede af overfladen. Findes i laboratorier verden over, AFM'er er arbejdshesteinstrumenter med forskellige anvendelser inden for videnskab og teknik.
Kun få HR-AFM'er findes i USA. Den ene er placeret ved Center for Functional Nanomaterials (CFN) - et US Department of Energy (DOE) Office of Science User Facility ved Brookhaven National Laboratory. I flere år har fysiker Percy Zahl fra CFN Interface Science and Catalysis Group opgraderet og tilpasset CFN HR-AFM hardware og software, hvilket gør det nemmere at betjene og erhverve billeder. Som højt specialiserede instrumenter kræver HR-AFM'er ekspertise at bruge. De fungerer ved meget lav temperatur (lige over det, der kræves for at gøre helium flydende). Desuden afhænger HR-billeddannelse af at fange et enkelt kuliltemolekyle på enden af spidsen.
Hvor udfordrende det end kan være at forberede og betjene instrumentet til eksperimenter, er det kun begyndelsen at se, hvordan molekyler ser ud. Dernæst skal billederne analyseres og fortolkes. Med andre ord, hvordan korrelerer billedtræk med molekylers kemiske egenskaber?
Sammen med teoretikere fra CFN og universiteter i Spanien og Schweiz stillede Zahl netop dette spørgsmål til hydrogenbundne netværk af trimesinsyre (TMA) molekyler på en kobberoverflade. Zahl begyndte at afbilde disse porøse netværk - lavet af kulstof, brint og oxygen - for et par år siden. Han var interesseret i deres potentiale til at begrænse atomer eller molekyler, der er i stand til at være vært for elektronspintilstande til kvanteinformationsvidenskab (QIS) applikationer. Men med eksperiment og grundlæggende simuleringer alene kunne han ikke forklare deres grundlæggende struktur i detaljer.
"Jeg havde mistanke om, at TMA-molekylernes stærke polaritet (ladningsområder) lå bag det, jeg så på AFM-billederne," sagde Zahl. "Men jeg havde brug for mere præcise beregninger for at være sikker."
I AFM måles den samlede kraft mellem probespidsen og molekylet. For et præcist match mellem eksperiment og simulering skal der dog tages højde for hver enkelt kraft i spillet. Grundlæggende modeller kan simulere kortrækkende kræfter for simple ikke-polære molekyler, hvor elektriske ladninger er jævnt fordelt. Men for kemisk rige strukturer, som findes i polære molekyler som trimesinsyre, skal elektrostatiske kræfter (der stammer fra den elektroniske ladningsfordeling inde i molekylet) og van der Waals-kræfter (tiltrækning mellem molekyler) også tages i betragtning. For at simulere disse kræfter har forskerne brug for den nøjagtige molekylære geometri, der viser, hvordan atomer er placeret i alle tre dimensioner, og de nøjagtige ladningsfordelinger inde i molekylerne. Gennem DFT-beregninger på Swiss National Supercomputing Center afslappede Aliaksandr Yakutovich strukturelt ringen med seks TMA-molekyler på en kobberplade indeholdende 1.800 kobberatomer. Ved strukturel afslapning er en grundlæggende geometrisk eller strukturel model optimeret til at finde konfigurationen af atomer med den lavest mulige energi.
I denne undersøgelse analyserede Zahl arten af selvsamlingen af TMA-molekyler til honeycomb-lignende netværksstrukturer på en ren kobberkrystal. Zahl afbildede oprindeligt strukturerne i stor skala med et scanning tunneling mikroskop (STM). Dette mikroskop scanner en metallisk spids over en overflade, mens der påføres en elektrisk spænding mellem dem. For at identificere, hvordan netværksstrukturen var på linje med substratet, bombarderede CFN-materialeforsker Jurek Sadowski prøven med lavenergielektroner og analyserede mønstret af diffrakterede elektroner. Endelig udførte Zahl HR-AFM, som er følsom over for højden af overfladetræk på en submolekylær skala.
"Med STM kan vi se netværkene af TMA-molekyler, men kan ikke let se orienteringen af kobber på samme tid," sagde Zahl. "Lavenergielektrondiffraktion kan fortælle os, hvordan kobber- og TMA-molekylerne er orienteret i forhold til hinanden. AFM giver os mulighed for at se den detaljerede kemiske struktur af molekylerne. Men for at forstå disse detaljer skal vi modellere systemet og bestemme præcist hvor atomerne i TMA-molekylerne sidder på kobber."
Til denne modellering brugte holdet tæthedsfunktionel teori (DFT) til at beregne de mest energisk gunstige arrangementer af TMA-molekyler på kobber. Ideen bag DFT er, at den samlede energi i et system er en funktion af dets elektrontæthed, eller sandsynligheden for at finde en elektron på et bestemt sted omkring et atom. Flere elektronegative atomer (som oxygen) har en tendens til at trække elektroner væk fra mindre elektronegative atomer (som kulstof og brint), de er bundet til, svarende til en magnet. Sådanne elektrostatiske interaktioner er vigtige for at forstå kemisk reaktivitet.
Mark Hybertsen, leder af CFN Theory and Computation Group, udførte indledende DFT-beregninger for et individuelt TMA-molekyle og to TMA-molekyler forbundet af hydrogenbindinger (en dimer). Aliaksandr Yakutovich fra [email protected] Laboratory of the Swiss Federal Laboratories for Materials Science and Technology (Empa) kørte derefter DFT-beregninger af et større TMA-netværk bestående af en komplet ring af seks TMA-molekyler.
Disse beregninger viste, hvordan molekylernes indre carbonring er forvrænget fra en hexagonal til en trekantet form i AFM-billedet på grund af stærke polariseringer forårsaget af tre carboxylgrupper (COOH). Derudover trækkes eventuelle ubundne iltatomer en smule ned mod overfladen af kobberatomer, hvor flere elektroner opholder sig. De beregnede også styrken af de to hydrogenbindinger, der dannes mellem to TMA-molekyler. Disse beregninger viste, at hver binding var omkring dobbelt så stærk som en typisk enkelt brintbinding.
"Ved at forbinde modeller i atomare skala til AFM-billeddannelseseksperimenterne kan vi forstå grundlæggende kemiske træk i billederne," sagde Hybertsen.
"Denne evne kan hjælpe os med at identificere kritiske molekyleegenskaber, herunder reaktivitet og stabilitet, i komplekse blandinger (såsom petroleum) baseret på HR-AFM-billeder," tilføjede Zahl.

En sammenligning mellem eksperimentelle (øverst) og simulerede (nederste tre ved forskellige probespids-prøvehøjder) AFM-billeder af to hydrogenbundne TMA-molekyler. Kredit:Brookhaven National Laboratory
For at lukke sløjfen mellem modellering og eksperiment indsatte samarbejdspartnere i Spanien DFT-resultaterne i en beregningskode, de udviklede til at generere simulerede AFM-billeder. Disse billeder passede perfekt til de eksperimentelle billeder.
"Disse nøjagtige simuleringer afslører det subtile samspil mellem den oprindelige molekylære struktur, deformationer induceret af interaktionen med substratet og de iboende kemiske egenskaber af molekylet, der bestemmer den komplekse, slående kontrast, som vi observerer i AFM-billederne," sagde Ruben Perez af Universidad Autónoma de Madrid.
Fra deres kombinerede tilgang viste holdet også, at linjelignende træk, der optræder mellem molekyler i AFM-billeder af TMA (og andre molekyler), ikke er fingeraftryk af hydrogenbindinger. De er snarere "artefakter" fra bøjning af AFM-sondemolekylet.
"Selvom hydrogenbinding er meget stærk for TMA-molekyler, er hydrogenbindinger usynlige i eksperimentet og simuleringen," sagde Zahl. "Det, der er synligt, er tegn på stærk elektrontilbagetrækning af carboxylgrupperne."
Dernæst planlægger Zahl at fortsætte med at studere dette modelsystem til netværks-selvsamling for at udforske dets potentiale for QIS-applikationer. Han vil bruge et nyt STM/AFM-mikroskop med yderligere spektroskopiske egenskaber, såsom dem til at kontrollere prøver med et magnetfelt og anvende radiofrekvente felter til prøver og karakterisere deres respons. Disse egenskaber vil gøre det muligt for Zahl at måle kvantespin-tilstandene for brugerdefinerede molekyler arrangeret i et perfekt array for at danne potentielle kvantebits.
Forskningen blev offentliggjort i Nanoscale . + Udforsk yderligere
Team måler opdelingen af en enkelt kemisk binding
 Varme artikler
Varme artikler
-
 pH-følsomme iridiumkomplekser som katalytiske anticancerforbindelserKredit:Ana C. Carrasco Kemoterapi er defineret som brugen af kemikalier til at nå og beskadige kræftceller. På vej mod tumoren, stofferne kan påvirke raske celler, såvel. For eksempel, cisplatin
pH-følsomme iridiumkomplekser som katalytiske anticancerforbindelserKredit:Ana C. Carrasco Kemoterapi er defineret som brugen af kemikalier til at nå og beskadige kræftceller. På vej mod tumoren, stofferne kan påvirke raske celler, såvel. For eksempel, cisplatin -
 Er de nuværende vandbehandlingsmetoder tilstrækkelige til at fjerne skadelige konstruerede nanopar…Kredit:Mary Ann Liebert, Inc., forlag Den øgede brug af manipulerede nanopartikler (ENMer) i kommercielle og industrielle applikationer vækker bekymring over miljø- og sundhedsvirkningerne af nano
Er de nuværende vandbehandlingsmetoder tilstrækkelige til at fjerne skadelige konstruerede nanopar…Kredit:Mary Ann Liebert, Inc., forlag Den øgede brug af manipulerede nanopartikler (ENMer) i kommercielle og industrielle applikationer vækker bekymring over miljø- og sundhedsvirkningerne af nano -
 Løsning af en naturlig gåde med vandfiltreringDisse foldekanaler hjælper med at transportere vand, mens de blokerer for uønskede molekyler som salt. Kredit:University of Texas ved Austin/Cockrell School of Engineering. For mange ingeniører og
Løsning af en naturlig gåde med vandfiltreringDisse foldekanaler hjælper med at transportere vand, mens de blokerer for uønskede molekyler som salt. Kredit:University of Texas ved Austin/Cockrell School of Engineering. For mange ingeniører og -
 DNA -nanoswitches afslører, hvordan livsmolekyler forbinderEt komplekst samspil mellem molekylære komponenter styrer næsten alle aspekter af biologiske videnskaber - sund organismeudvikling, sygdomsprogression, og lægemiddeleffektivitet afhænger alle af den m
DNA -nanoswitches afslører, hvordan livsmolekyler forbinderEt komplekst samspil mellem molekylære komponenter styrer næsten alle aspekter af biologiske videnskaber - sund organismeudvikling, sygdomsprogression, og lægemiddeleffektivitet afhænger alle af den m
- Hvilke robotter bruges i dag?
- Observerer nano-bio-interaktioner i realtid
- Højere temperatur, kraftigere regn
- Ledende natur i krystalstrukturer afsløret ved forstørrelse på 10 millioner gange
- Hvad er taktil stimulering?
- Forestil dig dette SELFI:NASA avancerer instrument til at studere Enceladus's fjer