


Prioner forklaret:Hvordan fejlfoldede proteiner driver fatale neurodegenerative sygdomme

Da bønderne i det 20. århundrede først observerede en uforklarlig neurologisk sygdom hos får, kaldte de den scrapie. Årtier senere opdagede videnskabsmænd, at synderen var et fejlfoldet protein - nu kendt som en prion - i stand til at fremkalde dødelige hjernesygdomme hos både dyr og mennesker.
TL;DR
Prioner er unormale proteinkonformationer, der kan replikere og forårsage dødelige neurodegenerative sygdomme såsom kogalskab, Creutzfeldt-Jakobs sygdom og kronisk svindsygdom.
Hvad er prioner?
Prioner er proteinholdige infektiøse partikler, der mangler nukleinsyrer. De opstår, når et normalt cellulært protein (ofte kaldet PrP C ) folder til en sygdomsfremkaldende konformation (PrP Sc ). Den forkert foldede form er rig på beta-sheetstruktur, hvorimod det sunde protein overvejende er alfa-helisk. Dette strukturelle skift gør det muligt for prion at skabe maling for fejlfoldning af andre normale proteiner, hvilket fører til en kaskade af skader.
Hvordan prioner replikeres
I modsætning til vira eller bakterier formerer prioner sig uden DNA eller RNA. Det patogene protein inducerer konformationelle ændringer i sunde proteiner og omdanner dem til den forkert foldede form. Denne autokatalytiske proces gør det muligt for prioner at formere sig uafhængigt af genetisk materiale - et fænomen, der fortsat er genstand for intens forskning.
Prionsygdomme hos mennesker og dyr
Hos mennesker omfatter prionlidelser:
- Creutzfeldt-Jakobs sygdom (CJD)
- Gerstmann-Straussler-Scheinkers syndrom
- Dødelig familiær søvnløshed
- Kuru
Hos dyr omfatter prionsygdomme:
- Bovin spongiform encephalopati (BSE) – almindeligvis kendt som kogalskab
- Scrapie hos får
- Kronisk spildsygdom (CWD) hos hjorte og elge
- Forskellige encephalopatier hos andre arter
Alle disse tilstande er progressive, neurodegenerative og i øjeblikket uhelbredelige. Symptomer involverer typisk demens, motorisk dysfunktion og til sidst død.
Forskningsbureauer såsom CDC og WHO vedligeholde ajourførte data om prionudbrud og forskningsfremskridt.
Selvom der ikke findes nogen definitive behandlinger, fokuserer igangværende undersøgelser på tidlig påvisning, terapeutiske antistoffer og nye småmolekylehæmmere, der kan afbryde fejlfoldningskaskaden.
 Varme artikler
Varme artikler
-
 Forskere identificerer en måde at svække malariaparasitter mod populær lægemiddelbehandlingKredit:CDC Indiana University School of Medicine -forskere har identificeret en måde at blokere parasiternes evne, der får malaria til at beskytte sig mod lægemiddelbehandlinger hos inficerede mus
Forskere identificerer en måde at svække malariaparasitter mod populær lægemiddelbehandlingKredit:CDC Indiana University School of Medicine -forskere har identificeret en måde at blokere parasiternes evne, der får malaria til at beskytte sig mod lægemiddelbehandlinger hos inficerede mus -
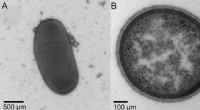 Ny bakterieart opdaget i tarmenMikrografierne af den elektronmikroskopiske analyse udført af Dr. Urska Repnik ved Kiel Universitys Central Microscopy Facility viser en af de nyopdagede bakterier (A) som en hel bakterie, der illus
Ny bakterieart opdaget i tarmenMikrografierne af den elektronmikroskopiske analyse udført af Dr. Urska Repnik ved Kiel Universitys Central Microscopy Facility viser en af de nyopdagede bakterier (A) som en hel bakterie, der illus -
 Regulering af frugtudvikling og modning ved DNA-methyleringFænotyper af støvdrager i SlCMT4 mutantlinjer. Kredit:Nanjing Agricultural University For nylig afslørede forskere fra Shanxi Datong University og Chongqing University, at SlCMT4, som koder for en
Regulering af frugtudvikling og modning ved DNA-methyleringFænotyper af støvdrager i SlCMT4 mutantlinjer. Kredit:Nanjing Agricultural University For nylig afslørede forskere fra Shanxi Datong University og Chongqing University, at SlCMT4, som koder for en -
 Pesticider kan få humler til at miste deres sus, undersøgelse finderBier. Kredit:University of Stirling Pesticider reducerer betydeligt antallet af pollenkorn, en humle er i stand til at indsamle, en ny undersøgelse fra University of Stirling har fundet. Forsknin
Pesticider kan få humler til at miste deres sus, undersøgelse finderBier. Kredit:University of Stirling Pesticider reducerer betydeligt antallet af pollenkorn, en humle er i stand til at indsamle, en ny undersøgelse fra University of Stirling har fundet. Forsknin
- Bærerne til energi og høje elektroner under glykolyse er?
- Højhastighedskameraundersøgelse viser, hvorfor kogende dråber tager fart
- Hvilke skyer er lave til jorden?
- Sådan beregnes en sum af kvadratiske afvigelser fra gennemsnittet (Summen af kvadrater)
- Hvordan dannes kuldioxid?
- Hvad er den type rock, som oceaniske plader er lavet af?