


Team udvikler matematiske teknikker til at forbedre beregningseffektiviteten inden for kvantekemi
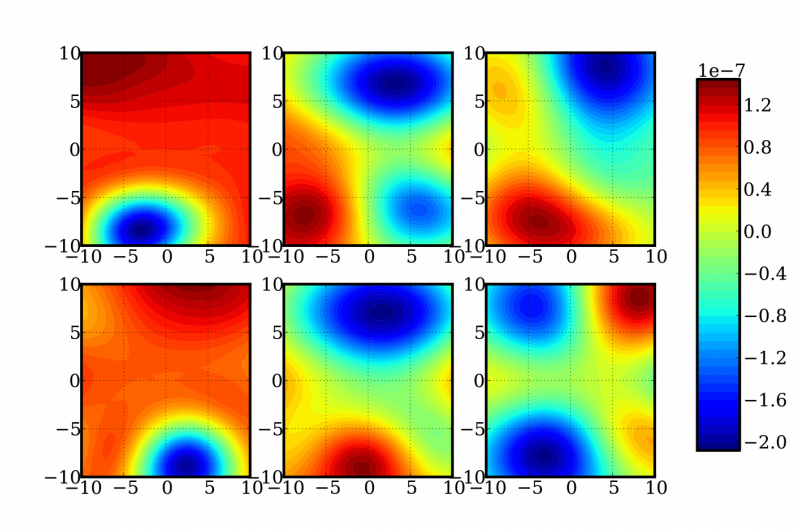
En skildring af tilfældige todimensionale skiver af en 12-dimensionel funktion til bestemmelse af energi- og frekvenskorrektioner af et formaldehydmolekyle. Kredit:Sandia National Laboratories
Forskere ved Sandia National Laboratories har udviklet nye matematiske teknikker til at fremme studiet af molekyler på kvante niveau.
Matematiske og algoritmiske udviklinger langs disse linjer er nødvendige for at muliggøre en detaljeret undersøgelse af komplekse kulbrintemolekyler, der er relevante ved motorforbrænding.
Eksisterende metoder til at tilnærme potentielle energifunktioner på kvanteskalaen har brug for for meget computerkraft og er dermed begrænset til små molekyler. Sandia -forskere siger, at deres teknik vil fremskynde kvantemekaniske beregninger og forbedre forudsigelser fra teoretiske kemimodeller. I betragtning af den hurtige beregning, disse metoder kan potentielt anvendes på større molekyler.
Sandia postdoktorforsker Prashant Rai arbejdede sammen med forskerne Khachik Sargsyan og Habib Najm ved Sandias forbrændingsforskningsfacilitet og samarbejdede med kvantekemikere So Hirata og Matthew Hermes ved University of Illinois i Urbana-Champaign. Beregningsenergi ved færre geometriske arrangementer end normalt krævet, teamet udviklede beregningsmæssigt effektive metoder til at tilnærme potentielle energioverflader.
En præcis forståelse af potentielle energioverflader, nøgleelementer i stort set alle beregninger af kvantedynamik, er nødvendig for nøjagtigt at estimere energien og frekvensen af molekylers vibrationstilstande.
"Hvis vi kan finde molekylets energi til alle mulige konfigurationer, vi kan bestemme vigtige oplysninger, såsom stabile tilstande af molekylær overgangsstruktur eller mellemliggende tilstande af molekyler i kemiske reaktioner, "Sagde Rai.
De første resultater af denne forskning blev offentliggjort i Molekylær fysik i en artikel med titlen "Low-rank canonical-tensor decomposition of potential energy overflates:application to grid-based diagrammatic vibrational Green's function theory."

Sandia National Laboratories forskere Prashant Rai, venstre, Habib Najm, centrum, og Khachik Sargsyan diskuterer matematiske teknikker, der bruges til at studere adfærden hos store molekyler i kvanteskala. Kredit:Dino Vournas
"Tilnærmelse af større molekylers potentielle energioverflader er en ekstremt udfordrende opgave på grund af den eksponentielle stigning i information, der kræves for at beskrive dem med hvert ekstra atom i systemet, "Sagde Rai." I matematik, det betegnes dimensionens forbandelse. "
Slog forbandelsen
Nøglen til at slå forbandelsen over dimensionalitet er at udnytte egenskaberne ved den specifikke struktur af de potentielle energioverflader. Rai sagde, at denne strukturinformation derefter kan bruges til at tilnærme de nødvendige højdimensionelle funktioner.
"Vi gør brug af det faktum, at selvom potentielle energioverflader kan være højdimensionelle, de kan godt tilnærmes som en lille sum af produkter med endimensionelle funktioner. Dette er kendt som den laveste struktur, hvor rangen af den potentielle energioverflade er antallet af udtryk i summen, "Rai sagde." En sådan antagelse om struktur er ret generel og er også blevet brugt i lignende problemer på andre områder. Matematisk, intuitionen af tilnærmelsesteknikker med lav rang kommer fra multilinjær algebra, hvor funktionen fortolkes som en tensor og nedbrydes ved hjælp af standard tensor-dekomponeringsteknikker. "
Energi- og frekvenskorrektionerne er formuleret som integraler af disse højdimensionelle energifunktioner. Tilnærmelse i et så lavt format gør disse funktioner let integrerbare, da det bryder integrationsproblemet til summen af produkter fra en- eller todimensionale integraler, så standard integrationsmetoder gælder.
Teamet afprøvede deres beregningsmetoder på små molekyler som vand og formaldehyd. Sammenlignet med den klassiske Monte Carlo -metode, den tilfældighedsbaserede standard arbejdshest til højdimensionelle integrationsproblemer, deres tilgang forudsagde energi og hyppighed af vandmolekyle, der var mere præcise, og det var mindst 1, 000 gange mere beregningsmæssigt effektiv.
Rai sagde, at det næste trin er at forbedre teknikken yderligere ved at udfordre den med større molekyler, såsom benzen.
"Tværfaglige undersøgelser, såsom kvantekemi og forbrændingsteknik, give muligheder for krydsbestøvning af ideer, derved give et nyt perspektiv på problemer og deres mulige løsninger, "Rai sagde." Det er også et skridt i retning af at bruge de seneste fremskridt inden for datavidenskab som en søjle for videnskabelig opdagelse i fremtiden. "
 Varme artikler
Varme artikler
-
 Simuleringer undersøger materialers ydeevne i NIF-eksperimenterDisse billeder viser beregnet lasereffekt pr. arealenhed på kapseloverfladen, der blev brugt i eksperimenterne. De sorte prikker angiver pegningen på kapseloverfladen. Kredit:Lawrence Livermore Nation
Simuleringer undersøger materialers ydeevne i NIF-eksperimenterDisse billeder viser beregnet lasereffekt pr. arealenhed på kapseloverfladen, der blev brugt i eksperimenterne. De sorte prikker angiver pegningen på kapseloverfladen. Kredit:Lawrence Livermore Nation -
 Magnetiske monopoler får en akustisk debutKredit:University of Oxford University College Cork (UCC) og University of Oxford Professor i fysik, Séamus Davis, har ledet et team af eksperimentelle fysikere i opdagelsen af den magnetiske st
Magnetiske monopoler får en akustisk debutKredit:University of Oxford University College Cork (UCC) og University of Oxford Professor i fysik, Séamus Davis, har ledet et team af eksperimentelle fysikere i opdagelsen af den magnetiske st -
 Måling af ændringer i magnetisk rækkefølge for at finde måder at overskride konventionel elektr…Pil angiver Mn3+ spins af sekskantet YMnO3, og rød stråle indikerer femtosekunds lysimpulser. Kredit:Tokyo Tech Forskere over hele verden leder konstant efter måder at forbedre eller overskride el
Måling af ændringer i magnetisk rækkefølge for at finde måder at overskride konventionel elektr…Pil angiver Mn3+ spins af sekskantet YMnO3, og rød stråle indikerer femtosekunds lysimpulser. Kredit:Tokyo Tech Forskere over hele verden leder konstant efter måder at forbedre eller overskride el -
 Spektral tilsløring kunne gøre objekter usynlige under realistiske forholdEn bredbåndsbølge belyser et objekt, som reflekterer grønt lys i det viste eksempel, gør objektet detekterbart af en observatør, der overvåger bølgen. En spektral usynlighedskappe forvandler den bloke
Spektral tilsløring kunne gøre objekter usynlige under realistiske forholdEn bredbåndsbølge belyser et objekt, som reflekterer grønt lys i det viste eksempel, gør objektet detekterbart af en observatør, der overvåger bølgen. En spektral usynlighedskappe forvandler den bloke
- Banebrydende flyvekontrolsystem undergår tredje testrunde
- Definition af karakteristika for lipidmolekyler
- Animal News Roundup! Tre underlige nye opdagelser, du har brug for at vide om
- Hvordan byer har sløvet modkulturens undergravende kraft
- Tidlig opdagelse af ozon på jordoverfladen kan hjælpe med at forhindre skader på druer og æbler
- Sådan beregnes en indvendig diameter