


Open source tilgang giver hurtigere, bedre opløselighedsforudsigelser
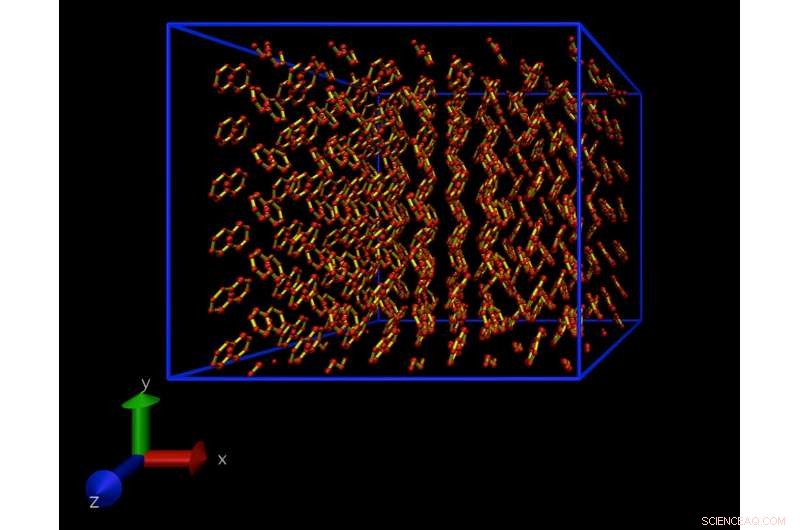
Et øjebliksbillede fra en Molecular Dynamics-simulering af en atomistisk model af en naphthalenkrystal. Denne krystal gentages periodisk i alle retninger, at eliminere overfladeeffekter. Kredit:Daan Frenkel, University of Cambridge
Opløseligheden af et givet stof - målet for, hvor godt stoffet opløses i et andet stof kaldet opløsningsmidlet - afhænger af grundlæggende egenskaber som temperatur og tryk, samt de kemiske identiteter af det opløste stof (det opløste) og opløsningsmidlet.
Forudsigelse af opløselighed er vigtig for en række anvendelser. På det farmaceutiske område, for eksempel, det er afgørende at kende et lægemiddels opløselighed, da det direkte bestemmer dets tilgængelighed for kroppen. Petroleumsindustrien giver et andet eksempel:Stoffer med lav opløselighed kan danne skæl eller uønskede aflejringer i rør eller på bor, forårsager blokeringer og andre store problemer.
På trods af vigtigheden af at forudsige opløselighed, det er ikke en nem sag. En tilgang, ved at bruge "brute force" simuleringer, kræver lange regnetider. Andre teknikker, mens hurtigere, undlader at forudsige nøjagtige opløselighedsværdier. Denne uge i Journal of Chemical Physics , forskere rapporterer om en ny type software, der muliggør praktiske opløselighedsestimater af stort set ethvert molekylært stof over brede temperatur- og trykområder. Koden gør brug af let tilgængelig open source-software og forventes at blive brugt bredt.
Daan Frenkel fra University of Cambridge i Storbritannien arbejdede sammen med kollegerne Lunna Li, også i Cambridge, og Tim Totton, af British Petroleum, at udvikle koden.
"Vi tog et bevidst valg om at bruge veldokumenterede, frit tilgængelig software, fordi vi ønskede at gøre vores tilgang tilgængelig for alle, " sagde Frenkel. "Et generelt værktøj til at beregne opløseligheder har manglet i lang tid. Den underliggende metode var der, men ingen havde faktisk lavet et fungerende program."
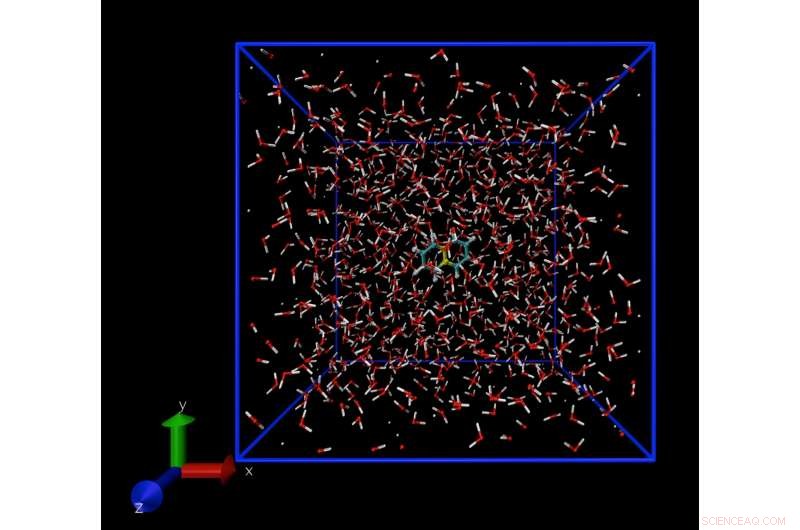
Et øjebliksbillede fra en Molecular Dynamics-simulering, der viser et enkelt naphthalenmolekyle, opløst i vand. Simuleringsteknikken gør det muligt at beregne koncentrationen af naphthalenmolekyler i vand ved opløselighedsgrænsen. Kredit:Daan Frenkel, University of Cambridge
Softwaren udviklet af denne gruppe bruger standard termodynamiske udtryk, der har været kendt siden midten af det 19. århundrede, såsom damptryk. Tilgangen udnytter det faktum, at når en fast eller flydende fase er i ligevægt, deres damptryk er lige store. Når en væske eller et fast stof opvarmes, molekyler undslipper og danner damp. Dette damptryk kan beregnes ved hjælp af computermodeller.
For eksempel, en sukkerklump, der opløses i vand:Sukkermolekyler eksisterer enten i fast tilstand - den krystallinske sukkerklump - eller helt omgivet af vandmolekyler, når de er opløst. Mængden af sukker i hver af de to faser, fast og opløsning, bestemmes af den energi, der kræves for at flytte sukkermolekyler mellem disse faser. Opløseligheden kan beregnes ved at beregne damptrykket af de to faser og sidestille dem.
For at modellere den faste fase, efterforskerne brugte en model, der omtales som en Einstein-krystal. I denne model, ikke-interagerende opløst stof molekyler er placeret på et gitter og bundet til et gitter punkt med en matematisk fjeder. Krystallens damptryk beregnes ved at beregne det nødvendige arbejde for at slukke for fjedrene og tænde for interaktioner mellem de forbundne molekyler.
For at modellere et opløst molekyle, efterforskerne brugte et standardenergipotentiale for det pågældende opløsningsmiddel, som var vand i eksemplerne brugt til at teste deres software, og beregnede arbejdet i tre trin. Først, der dannes et hulrum i opløsningsmidlet. Et opløst stof molekyle indsættes derefter i hulrummet og, endelig, hulrummet krympes til størrelsen af det opløste molekyle. Denne procedure eliminerer en række fejl og giver nøjagtige estimater af damptrykket og, dermed, opløseligheden.
I denne uges rapport, efterforskerne testede deres kode på naphthalen opløst i vand og forudsagde en opløselighed, der kan sammenlignes med eksperimentelle værdier. Fremtidige undersøgelser vil fokusere på at udvide softwaren, så den kan håndtere større opløste molekyler.
Sidste artikelLignende lipider klynges i sojabønnecellemembranmodel
Næste artikelNy diode har optisk styret kapacitans
 Varme artikler
Varme artikler
-
 Videnskabsprojekter om hvilken type chokolade, der smelter hurtigstEt videnskabsprojekt, der involverer chokolade, er en nem måde at lokke de studerende til at lære noget videnskabeligt, især hvis der er mulighed for at spise lidt chokolade undervejs. Smeltepunktet f
Videnskabsprojekter om hvilken type chokolade, der smelter hurtigstEt videnskabsprojekt, der involverer chokolade, er en nem måde at lokke de studerende til at lære noget videnskabeligt, især hvis der er mulighed for at spise lidt chokolade undervejs. Smeltepunktet f -
 Befugtning af overflader er overraskende svært at måle pålideligtVanddråbe på mikropiller. Kredit:Mika Latikka / Aalto University En gruppe forskere fra Aalto University i Finland og Sun Yat-sen University i Kina leverer en standardiseret tilgang til at forbedr
Befugtning af overflader er overraskende svært at måle pålideligtVanddråbe på mikropiller. Kredit:Mika Latikka / Aalto University En gruppe forskere fra Aalto University i Finland og Sun Yat-sen University i Kina leverer en standardiseret tilgang til at forbedr -
 Forudsiger, hvordan elektromagnetiske bølger interagerer med materialer på de mindste skalaerVenstre til højre:Yuanxun Ethan Wang, Tatsuo Itoh, Zhi Yao, og Rustu Umut Tok. Kredit:UCLA Samueli Engineering UCLA Samueli -ingeniører har udviklet et nyt værktøj til at modellere, hvordan magnet
Forudsiger, hvordan elektromagnetiske bølger interagerer med materialer på de mindste skalaerVenstre til højre:Yuanxun Ethan Wang, Tatsuo Itoh, Zhi Yao, og Rustu Umut Tok. Kredit:UCLA Samueli Engineering UCLA Samueli -ingeniører har udviklet et nyt værktøj til at modellere, hvordan magnet -
 En ny ikke-invasiv teknik til pergamentdiagnoseBillede af et pergamentmanuskript fra samlingen på Chartres-biblioteket (Copyright:CNRS-IRHT). Falske farvebilleder af ikke-lineær optisk mikroskopi med anden harmonisk generations signaler for det fi
En ny ikke-invasiv teknik til pergamentdiagnoseBillede af et pergamentmanuskript fra samlingen på Chartres-biblioteket (Copyright:CNRS-IRHT). Falske farvebilleder af ikke-lineær optisk mikroskopi med anden harmonisk generations signaler for det fi
- Oxfordshire Synchrotron
- Lær robotter at interagere med børn med autisme
- Nukleare strålingseffekter på planter
- NASA viser, at den største nedbør er fordrevet i tyfonen Chan-hom
- Kraftfuldt nyt kort viser miljøforringelse på tværs af Jorden
- For at forestille sig 5G-fremtiden, gense vores seneste trådløse fortid