


Forskere støber neurale net for at simulere molekylær bevægelse
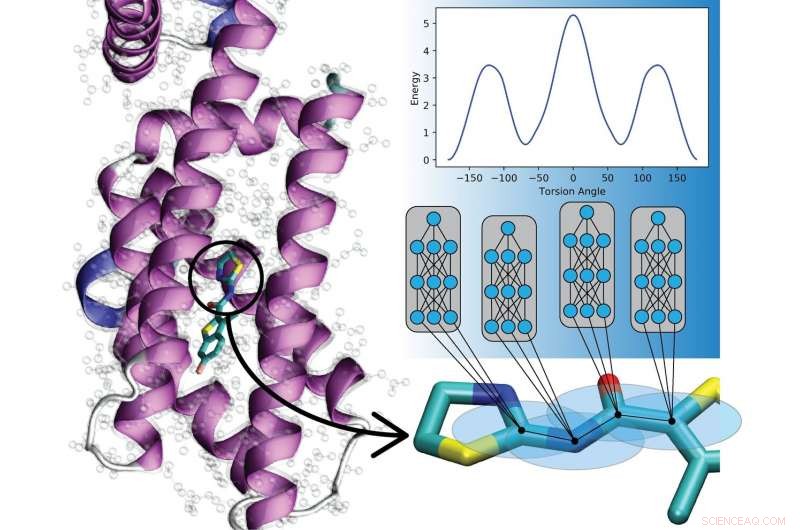
Nye deep learning -modeller forudsiger interaktionerne mellem atomer i organiske molekyler. Disse modeller vil hjælpe beregningsbiologer og forskere i lægemiddeludvikling med at forstå og behandle sygdom. Kredit:Los Alamos National Laboratory
Nyt arbejde fra Los Alamos National Laboratory, University of North Carolina at Chapel Hill, og University of Florida viser, at kunstige neurale net kan trænes til at kode kvantemekaniske love for at beskrive molekylers bevægelser, overladningssimuleringer potentielt på tværs af en lang række områder.
"Det betyder, at vi nu kan modellere materialer og molekylær dynamik milliarder af gange hurtigere i forhold til konventionelle kvantemetoder, med samme grad af nøjagtighed, "sagde Justin Smith, Los Alamos fysiker og Metropolis Fellow i laboratoriets teoretiske afdeling. At forstå, hvordan molekyler bevæger sig, er afgørende for at udnytte deres potentielle værdi for lægemiddeludvikling, proteinsimuleringer og reaktiv kemi, for eksempel, og både kvantemekanik og eksperimentelle (empiriske) metoder føder ind i simuleringerne.
Den nye teknik, kaldet ANI-1ccx potentialet, lover at fremme forskernes muligheder på mange områder og forbedre nøjagtigheden af maskinlæringsbaserede potentialer i fremtidige undersøgelser af metallegeringer og detonationsfysik.
Kvantemekaniske (QM) algoritmer, bruges på klassiske computere, kan præcist beskrive de mekaniske bevægelser af en forbindelse i dets driftsmiljø. Men QM skalerer meget dårligt med varierende molekylstørrelser, stærkt begrænsende omfanget af mulige simuleringer. Selv en lille stigning i molekylær størrelse inden for en simulering kan øge beregningsbyrden dramatisk. Så læger tyer ofte til at bruge empirisk information, som beskriver atomernes bevægelse i form af klassisk fysik og Newtons love, muliggør simuleringer, der skaleres til milliarder af atomer eller millioner af kemiske forbindelser.
Traditionelt set empiriske potentialer har måttet finde en afvejning mellem nøjagtighed og overførsel. Når potentialets mange parametre er finjusteret for en forbindelse, nøjagtigheden falder på andre forbindelser.
I stedet, Los Alamos -holdet, med University of North Carolina ved Chapel Hill og University of Florida, har udviklet en machine learning -tilgang kaldet transfer learning, der lader dem opbygge empiriske potentialer ved at lære af data indsamlet om millioner af andre forbindelser. Den nye tilgang med maskinindlæringens empiriske potentiale kan anvendes på nye molekyler i millisekunder, muliggøre forskning i et langt større antal forbindelser over meget længere tidsskalaer.
Sidste artikelHøj lysstyrke midt-infrarød laser udvider spektroskopisk analytisk tekniks horisont
Næste artikelSporing af mørkt stof
 Varme artikler
Varme artikler
-
 Fysikere beviser, at 2-D og 3-D væsker er fundamentalt forskelligeDette er et billede af atomare baner i en todimensionel væske, genereret af computersimuleringer. De fleste baner er aflange, og forlængelsen af tætte baner er ens. Dette er den visuelle signatur af
Fysikere beviser, at 2-D og 3-D væsker er fundamentalt forskelligeDette er et billede af atomare baner i en todimensionel væske, genereret af computersimuleringer. De fleste baner er aflange, og forlængelsen af tætte baner er ens. Dette er den visuelle signatur af -
 Sådan beregnes påvirkningskraftUnder en påvirkning konverteres energien fra et bevægeligt objekt til arbejde, og kraft spiller en vigtig rolle. For at oprette en ligning for styrken af enhver påvirkning, kan du indstille ligninge
Sådan beregnes påvirkningskraftUnder en påvirkning konverteres energien fra et bevægeligt objekt til arbejde, og kraft spiller en vigtig rolle. For at oprette en ligning for styrken af enhver påvirkning, kan du indstille ligninge -
 Hvordan interagerer Newtons bevægelseslove med tennis?Når du ser tennis eller enhver anden sport, ser du en demonstration af fysik, bare med mere jubel end det typiske fysikeksperiment. Centralt i handlingen er de tre bevægelseslove beskrevet i 1687 af S
Hvordan interagerer Newtons bevægelseslove med tennis?Når du ser tennis eller enhver anden sport, ser du en demonstration af fysik, bare med mere jubel end det typiske fysikeksperiment. Centralt i handlingen er de tre bevægelseslove beskrevet i 1687 af S -
 Lysbaseret computerhardware, der kan konkurrere med siliciumKredit:CC0 Public Domain Et team af forskere ved NTT Corporation har udviklet en måde at bruge lysbaseret computerhardware, der gør det muligt at konkurrere med silicium. I deres papir offentliggj
Lysbaseret computerhardware, der kan konkurrere med siliciumKredit:CC0 Public Domain Et team af forskere ved NTT Corporation har udviklet en måde at bruge lysbaseret computerhardware, der gør det muligt at konkurrere med silicium. I deres papir offentliggj
- Klimaindsatsen vil forbedre sundheden og redde liv nu og i fremtiden
- Sensor giver ny tilgang til detektering af molekyler på siliciumoverflader
- Sådan magnetiserer Washers
- Facebook trækker på brugerdata for at hjælpe med at bekæmpe coronavirus
- Astronomer afslører solkoronaens magnetfelt
- Hvordan bygger jeg et teleskop derhjemme?