


Fremskynde beregninger, der afslører, hvordan elektroner interagerer i materialer
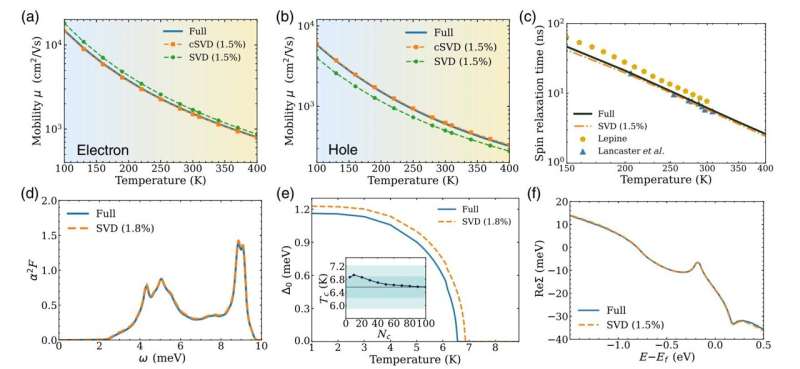
Materialeforskere og ingeniører vil gerne vide præcis, hvordan elektroner interagerer og bevæger sig i nye materialer, og hvordan de enheder, der er lavet med dem, vil opføre sig. Vil den elektriske strøm let flyde i materialet? Er der en temperatur, ved hvilken materialet bliver superledende, så strømmen kan flyde uden en strømkilde? Hvor længe vil kvantetilstanden af et elektronspin blive bevaret i nye elektroniske og kvanteenheder?
Et samfund af materialefysikere forsøger at løse sådanne spørgsmål ved at forstå, hvad der foregår inde i materialer, ved at beregne deres adfærd ned til niveauet for individuelle elektroninteraktioner og atombevægelser.
Nu har et Caltech-team gjort en nøgleopdagelse, der hjælper med at forenkle sådanne beregninger og fremskynde dem med en faktor på 50 eller mere, samtidig med at nøjagtigheden bevares. Som et resultat er det muligt at beregne elektroninteraktioner i mere komplekse materialer og enheder samt at udvikle nye beregninger, som man tidligere troede var umulige.
I et nyt papir offentliggjort i tidsskriftet Physical Review X , Caltechs Yao Luo, en kandidatstuderende i anvendt fysik; hans rådgiver Marco Bernardi, professor i anvendt fysik, fysik og materialevidenskab; og kolleger beskriver en ny datadrevet metode, der har muliggjort disse fremskridt. Deres tilgang forenkler de tætte beregningsmatricer, der bruges til at repræsentere de interaktioner, der finder sted i et materiale mellem elektroner og atomare vibrationer (eller fononer, som kan opfattes som individuelle enheder af vibrationsenergi).
Luo og Bernardi siger, at den nye metode tillader dem kun at bruge 1 til 2 % af de data, der typisk bruges til at løse sådanne problemer, hvilket i høj grad accelererer beregninger og afslører i processen de vigtigste interaktioner, der dikterer materialernes egenskaber.
"Dette var meget overraskende," siger Bernardi. "Elektron-fonon-interaktionerne beregnet med de komprimerede matricer er næsten lige så nøjagtige som den fulde beregning. Dette reducerer regnetiden og hukommelsesforbruget enormt med omkring to størrelsesordener i de fleste tilfælde. Det er også et elegant eksempel på Occams barbermaskine, idé om at favorisere simple fysiske modeller med minimalt antal parametre."
Find en ny mellemvej for feltet
Forskere inden for dette felt følger generelt en af to tilgange til at forstå materialer på dette mest grundlæggende niveau. En tilgang lægger vægt på at bygge minimale modeller, hvilket reducerer systemets kompleksitet, så forskerne kan justere en håndfuld parametre i pen-og-papir-beregninger for at få en kvalitativ forståelse af materialer.
Den anden begynder med intet andet end strukturen af et materiale og bruger såkaldte "første principper"-metoder - kvantemekaniske beregninger, der kræver store computere - til at studere materialers egenskaber med kvantitativ nøjagtighed.
Dette sidstnævnte sæt metoder, som Bernardis gruppe fokuserer på, bruger ekstremt store matricer med milliarder af indgange til at beregne elektroninteraktioner, der kontrollerer en lang række fysiske egenskaber. Det svarer til tusindvis af timers regnetid for hver beregning. Det nye arbejde antyder en slags mellemvej mellem de to tilgange, siger Bernardi.
"Med vores nye metode kan du afkorte størrelsen af disse matricer, udtrække nøgleinformationen og generere minimale modeller af interaktionerne i materialer."
Root ud af de vigtigste enkeltværdier
Hans gruppes tilgang er baseret på at anvende en metode kaldet singular value decomposition (SVD) på elektron-fonon-interaktionerne i et materiale. SVD-teknikken er meget udbredt inden for områder som billedkomprimering og kvanteinformationsvidenskab. Her giver det forfatterne mulighed for at adskille eller adskille de elektroniske og vibrationskomponenter i en matrix af tusinder eller millioner af elektron-fonon-interaktioner og tildele hver grundlæggende interaktion et nummer.
Disse reelle positive tal kaldes singulære værdier og rangerer de grundlæggende interaktioner i rækkefølge efter betydning. Så kan programmet eliminere alle på nær nogle få procent af interaktionerne i hver matrix, og kun efterlade de førende entalsværdier, en proces, der gør bestemmelsen billigere med en faktor, der er proportional med mængden af komprimering.
Så hvis programmet f.eks. kun beholder 1 % af singularværdierne, bliver beregningen hurtigere med en faktor 100. Forskerne har fundet ud af, at kun en lille brøkdel af singularværdierne, typisk 1 til 2 %, er det omtrentlige resultat bevarer næsten samme nøjagtighed som den fulde beregning.
"Ved at bruge SVD kan du skære antallet af enkeltværdier ned og kun fange hovedtrækkene i de matricer, der repræsenterer elektroniske interaktioner i et givet materiale," siger Luo, hovedforfatter på avisen, som er på sit tredje år i Bernardis gruppe.
"Dette afkorter den originale matrix og fremskynder dermed algoritmen og har den ekstra fordel, at det afslører, hvilke interaktioner i materialet der er dominerende."
Bernardi bemærker, at denne sidstnævnte fordel ved SVD-metoden giver forskerne en "fysisk intuition" om elektroninteraktioner i et materiale, noget der har manglet fra de første principberegninger tidligere. For eksempel, i en beregning, der involverede silicium, blev det klart, at den dominerende entalsværdi var forbundet med strækningen og klemningen af en bestemt binding.
"Det er noget simpelt, men før vi lavede beregningen, vidste vi ikke, at det var den stærkeste interaktion," forklarer Bernardi.
I papiret viser forskerne, at komprimeringen af matricer relateret til elektron-fonon-interaktioner ved hjælp af SVD-metoden giver nøjagtige resultater for forskellige egenskaber af materialer, som forskere måtte ønske at beregne, herunder ladningstransport, spinrelaksationstider og overgangstemperaturen for superledere .
Bernardi og hans team udvider de SVD-baserede beregninger til en bredere vifte af interaktioner i materialer og udvikler avancerede beregninger, som tidligere blev anset for umulige. Holdet arbejder også på at tilføje den nye SVD-metode til sin open source Perturbo-kode, en softwarepakke, der hjælper forskere med at beregne, hvordan elektroner interagerer og bevæger sig i materialer. Bernardi siger, at dette vil gøre det muligt for brugere i det videnskabelige samfund at forudsige materialeegenskaber forbundet med elektron-fonon-interaktioner betydeligt hurtigere.
Artiklen har titlen "Data-drevet komprimering af elektron-fonon-interaktioner." Sammen med Luo og Bernardi omfatter medforfattere på papiret kandidatstuderende Dhruv Desai (MS '22); Benjamin Chang (MS '20) og Jinsoo Park (Ph.D. '22), som nu er postdoc ved University of Chicago.
 Varme artikler
Varme artikler
-
 Ny tilgang til informationsoverførsel når kvantehastighedsgrænsenI en ny kvanteprotokol, grupper af kvantesammenfiltrede qubits (røde prikker) rekrutterer flere qubits (blå prikker) ved hvert trin for at hjælpe med hurtigt at flytte information fra et sted til et a
Ny tilgang til informationsoverførsel når kvantehastighedsgrænsenI en ny kvanteprotokol, grupper af kvantesammenfiltrede qubits (røde prikker) rekrutterer flere qubits (blå prikker) ved hvert trin for at hjælpe med hurtigt at flytte information fra et sted til et a -
 Holografiske metasurface-gassensorer til øjeblikkelige visuelle alarmerNumerisk optimering af asymmetrisk koblede metasurfaces. (A) Elementer af den foreslåede metasurface bestående af a-Si:H nanoantenner, der viser de elektriske og magnetiske feltintensitetsfordelinger
Holografiske metasurface-gassensorer til øjeblikkelige visuelle alarmerNumerisk optimering af asymmetrisk koblede metasurfaces. (A) Elementer af den foreslåede metasurface bestående af a-Si:H nanoantenner, der viser de elektriske og magnetiske feltintensitetsfordelinger -
 Ionisk beslutningstager, der er i stand til at lære selvFigur 1. Skematisk diagram af en ionisk enhed, der er i stand til at lære og træffe beslutninger ved hjælp af elektrokemiske fænomener induceret af hydrogenioners bevægelse. Figur 2. Den ioniske enhed
Ionisk beslutningstager, der er i stand til at lære selvFigur 1. Skematisk diagram af en ionisk enhed, der er i stand til at lære og træffe beslutninger ved hjælp af elektrokemiske fænomener induceret af hydrogenioners bevægelse. Figur 2. Den ioniske enhed -
 Kvanteteknik atomisk glatte enkeltkrystallinske sølvfilmSCULL (Single-crystalline Continuous Ultra-Smooth Low-loss Low-cost)-processen:To-trins afsætning af enkeltkrystallinske sølvfilm. (a) I det første trin, en AFT 2D. Ag (111) podekrystal aflejres under
Kvanteteknik atomisk glatte enkeltkrystallinske sølvfilmSCULL (Single-crystalline Continuous Ultra-Smooth Low-loss Low-cost)-processen:To-trins afsætning af enkeltkrystallinske sølvfilm. (a) I det første trin, en AFT 2D. Ag (111) podekrystal aflejres under
- Flere bevis på, at vejret i Californien er på vej mod ekstreme
- Hvad er en krystal og hvordan formes den?
- Langsom cykling er ikke kun for sjov – det er vigtigt for mange byarbejdere
- Nye modeller afslører indre kompleksitet af Saturn-månen
- Observatorium i høj højde kaster lys over oprindelsen af overskydende antistof
- Farveblindhedskorrigerende kontaktlinser