


Kemiker udvikler 3D-simuleringer af proteiner til spike i coronavirus
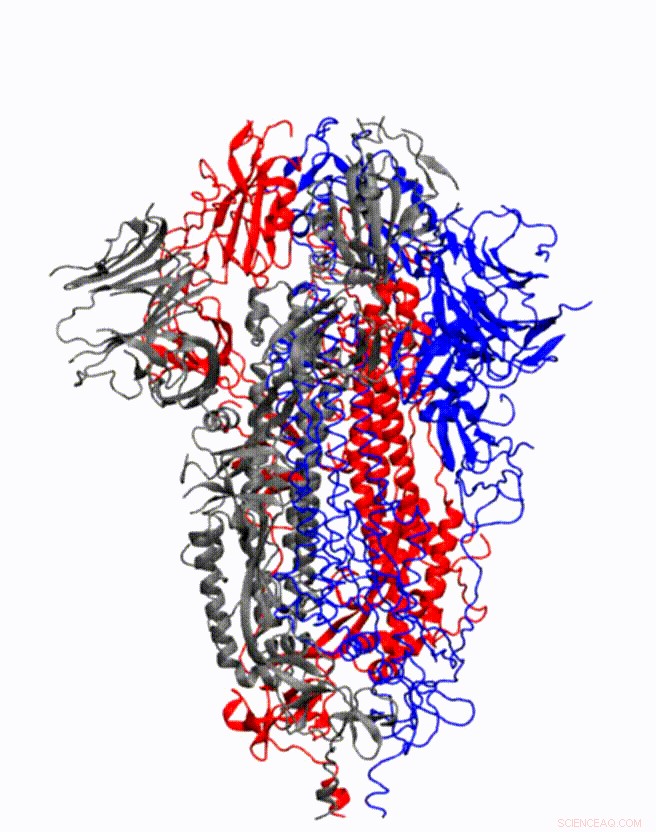
Animation, der viser, hvordan coronavirus -spike -protein ændrer form lige før det bindes til human celleceptor. Kredit:Illustration leveret af Mahmoud Moradi.
Beregningskemiker Mahmoud Moradi vil udvikle forbedret, 3D-simuleringer af molekylær dynamik i coronavirus spike glycoproteiner for at få en bedre forståelse af, hvordan virussen binder sig til menneskelige celler.
Kortlægning af, hvordan disse proteiner gennemgår konformationsændringer for at binde til værtscellereceptorer, er afgørende for udviklingen af coronavirus -vacciner og terapeutika. Simuleringer er især vigtige, fordi en ramme for lægemiddeldesign vil kræve dynamisk, tredimensionelle visualiseringer af cellestrukturer og adfærd, frem for et statisk billede.
"Som med andre vira, et afgørende trin i coronavirus -infektionsprocessen er viral indtræden, sagde Moradi, adjunkt ved J. William Fulbright College of Arts and Sciences. "Med coronavirus, vi ved, at disse pigglycoproteiner formidler adgang til den menneskelige celle. Både SARS-CoV-2, årsagen til COVID-19, og SARS-CoV, årsagen til SARS-epidemien 2002-2003, har piggproteiner, der binder sig til den samme receptor i humane celler. "
Moradis arbejde er en del af COVID-19 High Performance Computing Consortium, et samarbejde fra regeringen, industrien og akademiske partnere fokuserede på computerressourcer til COVID-19-forskning. I spidsen for Det Hvide Hus Kontor for Videnskab og Teknologi Politik, det amerikanske energiministerium og IBM, konsortiets frivillige gratis beregner tid og ressourcer på nogle af verdens mest magtfulde supercomputere.
For at udføre simuleringerne, Moradi har fået adgang til Frontera, en National Science Foundation-sponsoreret supercomputer, der ligger på University of Texas i Austin. Frontera er den største supercomputer på ethvert universitetsområde.
Moradis projekt drager fordel af flere nylige, højopløselige 3-D-modeller af coronavirus-spike-proteiner. Disse modeller kan bruges som indledende strukturer til at begynde simuleringer, der muliggør analyse af proteinernes detaljerede mekanismer og deres adfærd ved viral indgang. Forbedret, detaljerede simuleringer af en sådan molekylær dynamik vil give et komplet billede af proteiners strukturelle ændringer, samt hvordan de binder sig til angiotensin-konverterende enzym 2, den specifikke humane celle receptor.
Moradis forskning ligger i skæringspunktet mellem biologi, fysik, kemi, matematik, statistik og datalogi. Hans biomolekylære simuleringer og beregningsteorier forklarer, hvordan proteiner, arbejdshestens molekyler i celler, fungere på molekylært niveau. Hans arbejde forbedrer geometriske modeller for at beskrive, hvordan proteiner ændrer deres form, og hvordan disse ændringer påvirker et proteins adfærd. I februar, han modtog $ 650, 000 National Science Foundation Faculty Early Career Development -pris for dette arbejde.
 Varme artikler
Varme artikler
-
 Kemikere udvikler metode til hurtigt at screene, nøjagtigt identificere fentanyl og en bred vifte a…Kredit:McMaster University Forskere ved McMaster University har udviklet en ny lægemiddelscreeningsteknik, der kan føre til hurtig og nøjagtig identifikation af fentanyl, samt et stort antal andre
Kemikere udvikler metode til hurtigt at screene, nøjagtigt identificere fentanyl og en bred vifte a…Kredit:McMaster University Forskere ved McMaster University har udviklet en ny lægemiddelscreeningsteknik, der kan føre til hurtig og nøjagtig identifikation af fentanyl, samt et stort antal andre -
 Praktisk løsning til at forhindre korrosiv opbygning i nukleare systemerForskere har udtænkt en praktisk løsning til at forhindre ætsende ophobning i nukleare systemer. Billedet viser prøver af en standard reaktor zirconium legering med og uden vores CRUD-resistente belæg
Praktisk løsning til at forhindre korrosiv opbygning i nukleare systemerForskere har udtænkt en praktisk løsning til at forhindre ætsende ophobning i nukleare systemer. Billedet viser prøver af en standard reaktor zirconium legering med og uden vores CRUD-resistente belæg -
 Banebrydende materiale gør vej til brintbrug til brændselsceller under varme, tørre forholdForskere har udviklet en protonleder til brændselsceller baseret på polystyrenphosphonsyrer, der opretholder høj protonisk ledningsevne ved høje temperaturer uden vand. Kredit:Los Alamos National Labo
Banebrydende materiale gør vej til brintbrug til brændselsceller under varme, tørre forholdForskere har udviklet en protonleder til brændselsceller baseret på polystyrenphosphonsyrer, der opretholder høj protonisk ledningsevne ved høje temperaturer uden vand. Kredit:Los Alamos National Labo -
 Blandet halogenidkemi kan bruges til at kontrollere magnetisme i ultratynde magnetiske enhederChromchlorid og chrombromid var tidligere kendt som overgangsmetalhalogenider med magnetisering i planet og ud af planet. Boston College-forskere har opdaget en måde at fremstille blandede halogenider
Blandet halogenidkemi kan bruges til at kontrollere magnetisme i ultratynde magnetiske enhederChromchlorid og chrombromid var tidligere kendt som overgangsmetalhalogenider med magnetisering i planet og ud af planet. Boston College-forskere har opdaget en måde at fremstille blandede halogenider
- Fysikers undersøgelse viser, at silicium energihøstes
- Hvilken slags habitat lever elefanter i?
- Stoffer, der er uigennemtrængelige for vand
- Overvågning af ændringer i vådområdernes udstrækning kan hjælpe med at forudsige hastigheden a…
- Astronauter sigter mod isnende hjemkomst efter måneder i rummet
- Ohms lov: Hvad er det og hvorfor er det vigtigt?