


Ny genomisk metode afslører atomarrangementer af batterimateriale
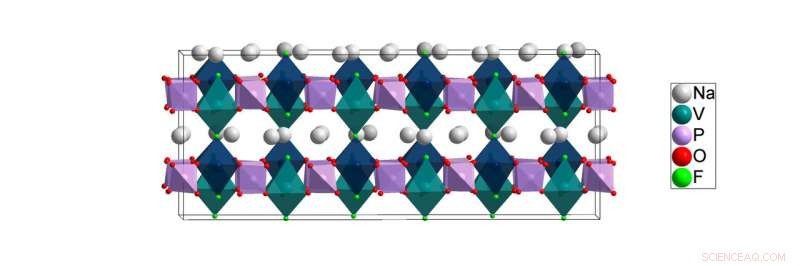
Lavtemperaturstrukturen af NVPF [Na3V2(PO4)2F3] løst i dette arbejde. Beregninger fra Lawrence Berkeley National Laboratory tyder på, at natriumatomerne (hvide) lettest kan bevæge sig i flyene mellem kationstederne for vanadium- (grøngrønne) og fosfor (lilla) atomer under batteribrug. Kredit:Brookhaven National Laboratory
Forskere ved det amerikanske energiministeriums (DOE) Brookhaven National Laboratory, Stony Brook University (SBU), Materials Project på DOE's Lawrence Berkeley National Laboratory (Berkeley Lab), University of California, Berkeley, og europæiske samarbejdspartnere har udviklet en ny måde at dechifrere materialers struktur på atomniveau baseret på data indsamlet fra malede pulverprøver. De beskriver deres tilgang og demonstrerer dens evne til at løse strukturen af et materiale, der viser lovende for shuttling af ioner gennem natrium-ion-batterier i et papir, der netop er offentliggjort i tidsskriftet Materialernes kemi .
"Vores tilgang kombinerer eksperimenter, teori, og moderne beregningsværktøjer til at levere de højkvalitets strukturelle data, der er nødvendige for at forstå vigtige funktionelle materialer, selv når kun pulverprøver er tilgængelige, " sagde den tilsvarende forfatter Peter Khalifah, der har en fælles ansættelse hos Brookhaven Lab og SBU.
Teknikken er på nogle måder en form for reverse engineering. I stedet for at løse strukturen direkte fra de eksperimentelle data målt på pulverprøven - et problem for komplekst til at være muligt for mange materialer - bruger den computeralgoritmer til at bygge og evaluere alle de plausible strukturer af et materiale. Ved at analysere "genomet" forbundet med et materiale på denne måde, det kan være muligt at finde den korrekte struktur, selv når denne struktur er så kompleks, at konventionelle metoder til strukturløsning mislykkes.
Batteri katode fryse-ramme
Til undersøgelsen beskrevet i papiret, Røntgenpulverdiffraktionseksperimenter blev udført ved ALBA synkrotronen i Barcelona, Spanien, af de europæiske samarbejdspartnere Matteo Bianchini og Francois Fauth, del af et team ledet af Christian Masquelier. Forskere brugte dette anlægs lyse røntgenstråler til at studere atomarrangementet af et natrium-ion batteri katodemateriale kendt som NVPF ved en række temperaturer lige fra stuetemperatur ned til de meget lave kryogene temperaturer, hvor atmosfæriske gasser bliver flydende. Dette arbejde er nødvendigt, fordi forstyrrelsen i stuetemperaturstrukturen af NVPF forsvinder, når den afkøles til kryogene temperaturer. Og mens batterierne kører nær stuetemperatur, at dechifrere materialets kryogene struktur er stadig kritisk vigtig, fordi kun denne lidelsesfri, lavtemperaturstruktur kan give forskerne en klar forståelse af den sande kemiske binding, der er til stede ved stuetemperatur. Dette kemiske bindingsmiljø har stor indflydelse på, hvordan ioner bevæger sig gennem strukturen ved stuetemperatur og påvirker dermed NVPF's ydeevne som batterimateriale.
"Bindingsmiljøet omkring natriumatomer - hvor mange naboer hver har - er stort set det samme ved lav temperatur, som det er ved stuetemperatur, " Khalifah forklarede, men at prøve at fange disse detaljer ved stuetemperatur er som at prøve at få børn til at sidde stille til et billede. "Alt bliver sløret, fordi ionerne bevæger sig for hurtigt rundt til at tillade et billede at blive taget." Af denne grund, nogle af bindingsmiljøerne udledt af rumtemperaturdata er ikke korrekte. I modsætning, kryogene temperaturer fryser natriumioners bevægelse for at give et retvisende billede af det lokale miljø, hvor natriumionerne sidder, når de ikke bevæger sig rundt.
"Når materialet afkøles, 24 tilstødende natriumioner er hver tvunget til at vælge et af to mulige steder, og deres foretrukne 'bestillingsmønster' med lavest energiforbrug kan løses, " sagde Khalifah.
En foreløbig analyse af pulverrøntgendiffraktionsdata af Bianchini indikerede, at bestillingsmønsteret er meget komplekst. For materialer med så komplekse bestillinger, det er typisk ikke muligt at løse deres tredimensionelle atomstruktur ved hjælp af pulverdiffraktionsdata.
"Pulverdiffraktionsdata bliver fladtrykt til én dimension, så en masse information går tabt, " sagde Khalifah.
Men materialer lavet af mange forskellige typer elementer, som det er tilfældet for NVPF - som er bygget af natriumatomer, vanadium, fosfor, fluor, og oxygen med en overordnet kemisk formel af Na 3 V 2 (PO 4 ) 2 F 3 -er for svære at vokse til større krystaller til mere konventionel 3-D røntgenkrystallografi.
Så, Brookhaven-gruppen samarbejdede med John Dagdelen og andre forskere ved Lawrence Berkeley National Laboratory for at udvikle en ny "genomisk" tilgang, der kan løse meget komplekse strukturer ved kun at bruge pulverdiffraktionsdata. Samarbejdet blev udført inden for Materialeprojektet, et DOE-finansieret forskerhold ledet af Kristin Persson ved LBNL, der udvikler innovative beregningsmetoder til at accelerere opdagelsen af nye funktionelle materialer.
"I stedet for at bruge pulverdiffraktionsdataene til direkte at løse strukturen, vi tog en alternativ tilgang, " sagde Khalifah. "Vi spurgte, "hvad er alle de plausible arrangementer af natriumioner i strukturen, ' og så testede vi hver af dem på en automatiseret måde for at sammenligne dem med de eksperimentelle data for at finde ud af, hvad strukturen var."
NVPF-strukturen er en af de mest komplekse, der nogensinde er løst for et materiale, der kun bruger pulverdiffraktionsdata.
"Vi kunne ikke have gjort denne videnskab uden moderne beregningsværktøjer - opregningsmetoderne, der blev brugt til at generere de kemisk plausible strukturer og de sofistikerede automatiserede scripts til at forfine de strukturer, der brugte pymatgen (Python Materials Genomics) softwarebiblioteket, " sagde Khalifah.
Nulstilling af strukturen
Baseret på den tilgængelige strukturelle viden for NVPF og på et sæt grundlæggende kemiske regler for binding, der er mere end en halv million plausible bestillingsmønstre for natriumatomerne i NVPF. Selv efter at have anvendt beregningsalgoritmer til at identificere ækvivalente strukturer genereret gennem forskellige bestillingsvalg, næsten 3, 000 unikke mulige bestillinger tilbage.
"Disse 3, 000 prøvestrukturer er mere, end det med rimelighed kan testes i hånden, men deres rigtighed kunne vurderes af en enkelt computer, der arbejder non-stop i omkring to dage, " sagde Khalifah.
Korrektheden af hver forsøgsstruktur blev evalueret ved hjælp af software til at forudsige, hvordan dens pulverrøntgendiffraktionsmønster ville se ud, og derefter sammenligne de beregnede resultater med de eksperimentelt målte diffraktionsdata, arbejde udført af Stony Brook Ph.D. elev Gerard Mattei. Hvis forskellen mellem de forudsagte og observerede diffraktionsmønstre er relativt lille, softwaren kan optimere enhver prøvestruktur ved at justere positionerne af dets konstituerende atomer for at forbedre overensstemmelsen mellem de beregnede og observerede mønstre.
Men selv efter sådanne justeringer, næsten 2, 500 af de optimerede strukturer kunne bruges til at passe de eksperimentelle diffraktionsdata godt.
"Vi havde ikke forventet at få så mange gode pasforme, " sagde Khalifah. "Så, vi havde en anden udfordring med at bestemme, hvilken af de mange mulige strukturer, der var korrekte ved at se på, hvilken der havde den korrekte symmetri."
Krystallografisk symmetri giver de regler, der begrænser, hvordan atomer kan arrangeres i et materiale, så fuld forståelse af symmetrien af en struktur er nødvendig for at beskrive den korrekt, Khalifah bemærkede.
Holdet havde genereret hver af forsøgsstrukturerne med et specifikt sæt symmetribegrænsninger. Og selvom det var meget udfordrende at bestemme den sande symmetri af en prøvestruktur efter dens optimering, en sammenligning af alle 2, 500 optimerede strukturer gjorde det muligt for forskerne at bestemme, hvilke symmetrielementer der var nødvendige for korrekt at beskrive den sande struktur af NVPF.
Evnen til at sammenligne resultater på tværs af mange forsøg giver en højere grad af tillid til den endelige løsning og er en yderligere fordel, som den nye metode, der anvendes i dette arbejde, har i forhold til traditionelle tilgange. Desuden, teoretiske beregninger udført af LBNL-forskerne John Dagdelen og Alex Ganose indikerede, at den endelige løsning er stabil mod forvrængninger, bekræfter gyldigheden af dette resultat.
Den løste struktur afslørede, at der er meget større diversitet i bindingen af natriumatomer, end det tidligere var blevet anerkendt.
"Fra rumtemperaturdataene, det viste sig på en misvisende måde, at alle natriumatomer var bundet til enten seks eller syv naboatomer, " sagde Khalifah. "I modsætning hertil, lavtemperaturdataene indikerede klart, at nogle natriumatomer har så få som fire naboer. Et resultat af dette er, at natriumatomerne med færre naboer er meget mindre låst på plads og forventes dermed at have lettere ved at bevæge sig gennem strukturen - en egenskab, der er afgørende for batterifunktionen."
Forfatterne mener, at denne nye tilgang bør være bredt anvendelig til at løse de komplekse strukturer, der almindeligvis forekommer i batterimaterialer, når ioner fjernes under opladning. Dette er især relevant i materialer, der anvendes i natrium- og kalium-ion-batterier, som er ved at blive udviklet som billigere og mere rigelige alternativer til lithium-ion batterimaterialer. Denne forskning bør derfor spille en vigtig rolle i at frigøre potentialet af jordrige materialer, der kan bruges til at opskalere energilagringskapaciteter for at imødekomme samfundsmæssige behov, såsom lagring i netskala.
Sidste artikelKøler rødglødende stål med varmt vand
Næste artikelForskere skaber hybridvævskonstruktion til bruskregenerering
 Varme artikler
Varme artikler
-
 Forskere bruger elektrostatisk ladning til at samle partikler til materialer, der efterligner ædels…Til venstre, små krystaller afbildes ved hjælp af et scanningselektronmikroskop, skelne mellem de enkelte byggesten, som består af kugleformede polystyrenperler. Til højre, større krystaller afbildes
Forskere bruger elektrostatisk ladning til at samle partikler til materialer, der efterligner ædels…Til venstre, små krystaller afbildes ved hjælp af et scanningselektronmikroskop, skelne mellem de enkelte byggesten, som består af kugleformede polystyrenperler. Til højre, større krystaller afbildes -
 Hydrodynamik i cellestudierKredit:IBM I samarbejde med ETH Zürich, vores team hos IBM Research – Zurich udgav en artikel, der gennemgår interaktionen mellem væskestrømme og biologiske celler. Vores arbejde blev vist på fors
Hydrodynamik i cellestudierKredit:IBM I samarbejde med ETH Zürich, vores team hos IBM Research – Zurich udgav en artikel, der gennemgår interaktionen mellem væskestrømme og biologiske celler. Vores arbejde blev vist på fors -
 Sådan opløses EDTA i WaterEthylendiaminetetraeddikesyre, eller EDTA, er en farveløs syre, som den amerikanske Food and Drug Administration har godkendt til behandling af bly- og tungmetalforgiftning, samt hypercalcæmi og ventr
Sådan opløses EDTA i WaterEthylendiaminetetraeddikesyre, eller EDTA, er en farveløs syre, som den amerikanske Food and Drug Administration har godkendt til behandling af bly- og tungmetalforgiftning, samt hypercalcæmi og ventr -
 Team -afkodet molekylær mekanisme, der hæmmer sværmende motilitet hos bakteriepopulationerDr. Thomas Boettcher. Kredit:University of Konstanz I naturen, bakterier forekommer mest i multicellulære kollektiver, frem for som enkeltpersoner. De er i stand til at koordinere deres adfærd, me
Team -afkodet molekylær mekanisme, der hæmmer sværmende motilitet hos bakteriepopulationerDr. Thomas Boettcher. Kredit:University of Konstanz I naturen, bakterier forekommer mest i multicellulære kollektiver, frem for som enkeltpersoner. De er i stand til at koordinere deres adfærd, me