


Omfattende elektroniske strukturmetoder til materialedesign
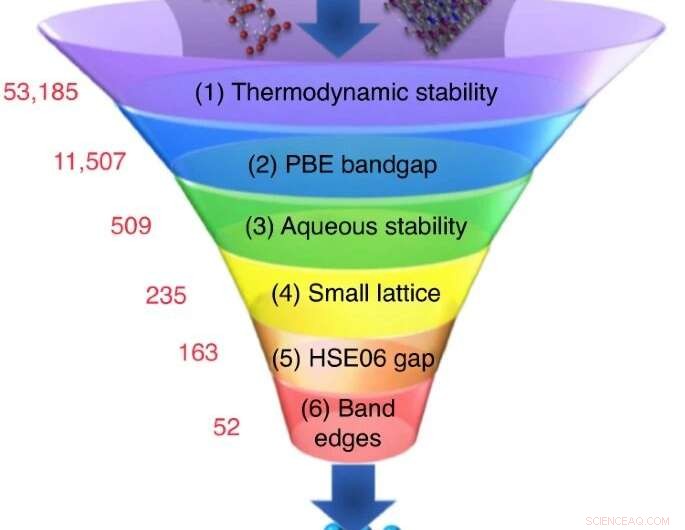
Et stort antal kandidatmaterialer er valgt fra eksperimentelle eller beregningsmæssige databaser, og en sekvens af screeningsberegninger reducerer deres antal ned til et lille sæt af kandidater med de mest lovende egenskaber. Kredit:Nicola Marzari
Nicola Marzari, leder af Theory and Simulation of Materials-laboratoriet på EFPL og direktør for NCCR MARVEL, har netop udgivet en anmeldelse af elektroniske strukturmetoder som en del af en specialudgave Insight on Computational Materials Design, udgivet af Naturmaterialer . Artiklen, skrevet med Andrea Ferretti fra CNR-Instituto Nanoscienze og Chris Wolverton fra Northwestern University, giver et overblik over disse metoder, diskuterer deres anvendelse til forudsigelse af materialeegenskaber, og undersøger forskellige strategier, der bruges til at målrette de bredere mål for materialedesign og -opdagelse. Ser frem til, forfatterne overvejer nye udfordringer i den forudsigelige nøjagtighed af beregningerne, og i at adressere den virkelige kompleksitet af materialer og enheder. De understreger også vigtigheden af de beregningsmæssige infrastrukturer, der understøtter sådan forskning, og hvordan planlægningen for finansiering af disse og de understøttende karrieremodeller kun lige er begyndt at dukke op.
I løbet af de sidste 20 år, simuleringer af de første principper er blevet kraftfulde, meget brugte værktøjer i mange, forskellige områder inden for videnskab og teknik. Fra nanoteknologi til planetarisk videnskab, fra metallurgi til kvantematerialer, de har fremskyndet identifikationen, karakterisering, og optimering af materialer enormt. De har ført til forbløffende forudsigelser - fra ultrahurtig termisk transport til elektron-fonon-medieret superledning i hydrider til fremkomsten af flade bånd i snoet dobbeltlagsgrafen - som har inspireret til bemærkelsesværdige eksperimenter.
Det nuværende fremstød for at supplere eksperimenter med simuleringer; fortsatte, hurtig vækst i computergennemstrømningskapacitet; maskinlærings og kunstig intelligenss evne til at fremskynde materialeopdagelse samt løftet om disruptive acceleratorer såsom kvanteberegning til eksponentielt dyre opgaver betyder, at det er tydeligt, at disse metoder vil blive stadig mere relevante som tiden går. Det er da et passende tidspunkt at gennemgå mulighederne såvel som begrænsningerne af de elektroniske strukturmetoder, der ligger til grund for disse simuleringer. Marzari, Ferretti og Wolverton behandler denne opgave i papiret "Elektroniske strukturmetoder til materialedesign, " netop offentliggjort i Naturmaterialer .
"Simuleringer mislykkes ikke på spektakulære måder, men kan subtilt skifte fra at være uvurderlige til knap nok gode til bare ubrugelige, " sagde forfatterne i avisen. "Årsagerne til fiasko er mange, fra at strække metodernes muligheder til at forsage kompleksiteten af rigtige materialer. Men simuleringer er også uerstattelige:De kan vurdere materialer under tryk- og temperaturforhold så ekstreme, at intet eksperiment på jorden er i stand til at replikere, de kan udforske med stadigt stigende smidighed det store rum af materialers faser og kompositioner i søgen efter det uhåndgribelige materialegennembrud, og de kan direkte identificere de mikroskopiske årsager og oprindelsen af en makroskopisk egenskab. Sidst, de deler et nøgleelement i forskningen med alle grene af computervidenskab:De kan gøres reproducerbare og åbne og delbare på måder, som ingen fysisk infrastruktur nogensinde vil være."
Forfatterne ser først på rammerne for tæthedsfunktionel teori (DFT) og giver et overblik over de stadig mere komplekse tilgange, der kan forbedre nøjagtigheden eller udvide omfanget af simuleringer. De diskuterer derefter de muligheder, som computermaterialevidenskab har udviklet til at udnytte denne værktøjskasse og levere forudsigelser for materialers egenskaber under realistiske forhold med stadigt stigende kompleksitet. Endelig, de fremhæver, hvordan fysik- eller datadrevne tilgange kan give rationelle, Høj gennemstrømning, eller kunstig intelligens veje til materialeopdagelse, og forklare, hvordan en sådan indsats ændrer hele forskningsøkosystemet.
Ser frem til, forfatterne siger, at udvikling af metoder, der kan vurdere den termodynamiske stabilitet, syntesebetingelser, fremstillingsevne, og tolerance af de forudsagte egenskaber over for iboende og ydre defekter i nye materialer vil være en væsentlig udfordring. Forskere skal muligvis udvide DFT-estimater med mere avancerede elektroniske strukturmetoder eller maskinlæringsalgoritmer for at forbedre nøjagtigheden, og bruge beregningsmetoder til at adressere realistiske forhold såsom vibrationsentropier, koncentrationen af defekter og anvendte elektrokemiske potentialer.
Endelig, i betragtning af den udvidede rolle, som sådanne metoder sandsynligvis vil spille i de kommende årtier, forfatterne bemærker, at støtte og planlægning af de nødvendige beregningsmæssige infrastrukturer - udbredt videnskabelig software, verifikation af koder og validering af teorier, formidling og kuration af beregningsdata, værktøjer og arbejdsgange samt de tilknyttede karrieremodeller, disse indebærer og kræver - er kun lige begyndt at dukke op.
 Varme artikler
Varme artikler
-
 Kuldetilpassede enzymer kan transformeres ved stuetemperaturKredit:CC0 Public Domain Enzymer fra kulde-elskende organismer, der lever ved lave temperaturer, tæt på vands frysepunkt, udviser meget karakteristiske egenskaber. I en ny undersøgelse offentliggj
Kuldetilpassede enzymer kan transformeres ved stuetemperaturKredit:CC0 Public Domain Enzymer fra kulde-elskende organismer, der lever ved lave temperaturer, tæt på vands frysepunkt, udviser meget karakteristiske egenskaber. I en ny undersøgelse offentliggj -
 Aktiv sigtning kunne forbedre dialyse- og vandrensningsfiltreFor at adskille molekyler bruges normalt et colander-filter (venstre side). En ny type filter, hvor hullerne i sien kan aktiveres udforskes. (Højre side) Takket være en ekstern strømindgang, hullerne
Aktiv sigtning kunne forbedre dialyse- og vandrensningsfiltreFor at adskille molekyler bruges normalt et colander-filter (venstre side). En ny type filter, hvor hullerne i sien kan aktiveres udforskes. (Højre side) Takket være en ekstern strømindgang, hullerne -
 Brugen af kulbrintegasKulbrinter er molekyler af kulstofbrint og ilt, der har forskellige kemiske og fysiske egenskaber afhængigt af strukturen for deres binding. Disse bindinger kan være enkle, multiple eller hexagonal
Brugen af kulbrintegasKulbrinter er molekyler af kulstofbrint og ilt, der har forskellige kemiske og fysiske egenskaber afhængigt af strukturen for deres binding. Disse bindinger kan være enkle, multiple eller hexagonal -
 Ledende natur i krystalstrukturer afsløret ved forstørrelse på 10 millioner gangeUniversity of Minnesota Professor K. Andre Mkhoyan og hans team brugte analytisk scanning transmissionselektronmikroskopi (STEM), som kombinerer billeddannelse med spektroskopi, at observere metallisk
Ledende natur i krystalstrukturer afsløret ved forstørrelse på 10 millioner gangeUniversity of Minnesota Professor K. Andre Mkhoyan og hans team brugte analytisk scanning transmissionselektronmikroskopi (STEM), som kombinerer billeddannelse med spektroskopi, at observere metallisk
- Socialrådgivere er ikke inkompetente børnesnabere - så hvorfor bliver de fremstillet på den måd…
- Fermi ser gammastråler fra skjulte soludbrud
- Tæmme vilde elektroner i grafen
- Et nyt system efterligner det menneskelige øjes fokusaktivitet
- PML bruger kombinerede optiske teknikker til at give vigtige svar på grafenstrukturer
- Bevarelse af synet for astronauter