


Forskere skaber ny AI-pipeline til at identificere molekylære interaktioner

At forstå, hvordan proteiner interagerer med hinanden, er afgørende for at udvikle nye behandlinger og forstå sygdomme. Takket være beregningsmæssige fremskridt har et team af forskere ledet af adjunkt i kemi Alberto Perez udviklet en algoritme til at identificere disse molekylære interaktioner.
Perez' forskerhold omfattede to kandidatstuderende fra UF, Arup Mondal og Bhumika Singh, og en håndfuld forskere fra Rutgers University og Rensselaer Polytechnic Institute. Holdet offentliggjorde deres resultater i Angewandte Chemie International Edition .
Dette innovative værktøj, som er navngivet AF-CBA Pipeline, tilbyder uovertruffen nøjagtighed og hastighed til at udpege de stærkeste peptidbindere til et specifikt protein. Det gør den ved at bruge kunstig intelligens til at simulere molekylære interaktioner, sortere gennem tusindvis af kandidatmolekyler for at identificere det molekyle, der interagerer bedst med proteinet af interesse.
Den AI-drevne tilgang tillader pipelinen at udføre disse handlinger på en brøkdel af den tid, det ville tage mennesker eller traditionelle fysikbaserede tilgange at udføre den samme opgave.
"Tænk på det som en købmand," forklarede Perez. "Når du vil købe den bedst mulige frugt, skal du sammenligne størrelser og aspekter. Der er selvfølgelig for mange frugter til at prøve dem alle sammen, så du sammenligner et par stykker, før du foretager et valg. Denne AI-metode kan dog ikke kun prøv dem alle, men kan også pålideligt udvælge den bedste."
Typisk er proteinerne af interesse dem, der forårsager mest skade på vores kroppe, når de opfører sig forkert. Ved at finde ud af, hvilke molekyler der interagerer med disse problematiske proteiner, åbner pipelinen veje for målrettede terapier til bekæmpelse af lidelser såsom inflammation, immunforstyrrelser og kræft.
"At kende strukturen af det stærkeste peptidbindemiddel hjælper os til gengæld med det rationelle design af nye lægemidler," sagde Perez.
Pipelinens banebrydende karakter forstærkes af dens fundament på allerede eksisterende teknologi:et program kaldet AlphaFold. AlphaFold er udviklet af Google Deepmind og bruger deep learning til at forudsige proteinstrukturer. Denne afhængighed af velkendt teknologi vil være en velsignelse for rørledningens tilgængelighed for forskere og vil hjælpe med at sikre dens fremtidige anvendelse.
Fremadrettet sigter Perez og hans team på at udvide deres pipeline for at få yderligere biologisk indsigt og hæmme sygdomsagenter. De har to vira i kikkerten:murin leukæmivirus og Kaposis sarkomvirus. Begge vira kan forårsage alvorlige helbredsproblemer, især tumorer, og interagere med hidtil ukendte proteiner.
"Vi ønsker at designe nye biblioteker af peptider," sagde Perez. "AF-CBA vil give os mulighed for at identificere de designede peptider, der binder stærkere end de virale peptider."
Flere oplysninger: Arup Mondal et al., A Computational Pipeline for Accurate Prioritering of Protein-Protein Binding Candidates in High-Throughput Protein Libraries, Angewandte Chemie International Edition (2024). DOI:10.1002/anie.202405767
Journaloplysninger: Angewandte Chemie International Edition
Leveret af University of Florida
 Varme artikler
Varme artikler
-
 Varme elektroner sender kuldioxid tilbage til fremtidenKatalysatornanopartiklerne udviklet af KAUST-forskere bruger lysenergi til at omdanne kuldioxid og brint til metan. Kredit:KAUST; Anastasia Serin Atmosfærisk kuldioxid (CO 2 ) er en væsentlig dr
Varme elektroner sender kuldioxid tilbage til fremtidenKatalysatornanopartiklerne udviklet af KAUST-forskere bruger lysenergi til at omdanne kuldioxid og brint til metan. Kredit:KAUST; Anastasia Serin Atmosfærisk kuldioxid (CO 2 ) er en væsentlig dr -
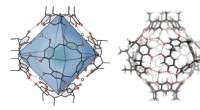 Ny tilgang til terpensynteserTil venstre, det omkring 1,4 kubik nanometer store hulrum i den molekylære kapsel er fremhævet med blåt. Til højre, samhørigheden af kapslen via hydrogenbindinger (grønne stiplede linjer) er synlig.
Ny tilgang til terpensynteserTil venstre, det omkring 1,4 kubik nanometer store hulrum i den molekylære kapsel er fremhævet med blåt. Til højre, samhørigheden af kapslen via hydrogenbindinger (grønne stiplede linjer) er synlig. -
 Forskere udvikler værktøj til at hjælpe med udvikling, effektiviteten af brintdrevne bilerSkematisk indre funktion af elektroderne i en brændselscelle, og vigtigheden af nøgleparametre. Kredit:Heinz et al., 2021 Udbredt anvendelse af brintdrevne køretøjer frem for traditionelle elekt
Forskere udvikler værktøj til at hjælpe med udvikling, effektiviteten af brintdrevne bilerSkematisk indre funktion af elektroderne i en brændselscelle, og vigtigheden af nøgleparametre. Kredit:Heinz et al., 2021 Udbredt anvendelse af brintdrevne køretøjer frem for traditionelle elekt -
 Maskinlæringsværktøj kan hjælpe med at udvikle hårdere materialerKredit:CC0 Public Domain For ingeniører, der udvikler nye materialer eller beskyttende belægninger, der er milliarder af forskellige muligheder for at sortere igennem. Labtest eller endda detaljer
Maskinlæringsværktøj kan hjælpe med at udvikle hårdere materialerKredit:CC0 Public Domain For ingeniører, der udvikler nye materialer eller beskyttende belægninger, der er milliarder af forskellige muligheder for at sortere igennem. Labtest eller endda detaljer
- Blå og guld-satellitter var på vej til Mars i 2024
- Sådan fodres en Mockingbird
- BAMS-rapport:Rekordhøje drivhusgasser, havniveauer i 2021
- Coca-Cola-aktier bobler op med højere overskud
- Lukker Elon Musk en besværlig Twitter-konto eller driller den? (Opdater)
- Quantum-dot spektrometer er lille nok til at fungere i en smartphone