


Elegant teori viser, hvordan vand hjælper med at adskille ioner involveret i materialesyntese og -fremstilling
Forskerholdet anvendte density functional theory (DFT) beregninger til at undersøge opførselen af vandmolekyler i nærvær af ioner. DFT er et kraftfuldt værktøj til at simulere den elektroniske struktur af materialer og molekyler. Holdet fokuserede på to specifikke systemer:opløsningen af lithiumioner (Li+) og samspillet mellem vandmolekyler og en lithium-ilt (Li-O) overflade.
I tilfælde af Li+-opløsning afslørede DFT-beregningerne, at vandmolekyler danner en højordnet hydreringsskal omkring lithium-ionen. Denne ordnede struktur er resultatet af de stærke elektrostatiske interaktioner mellem den positivt ladede lithiumion og de negativt ladede oxygenatomer i vandmolekyler. Hydratiseringsskallen beskytter effektivt lithiumionen mod at interagere med andre ioner eller molekyler i opløsningen, hvilket er afgørende for at stabilisere ionen og lette dens transport i elektrokemiske reaktioner.
Desuden undersøgte forskerne samspillet mellem vandmolekyler og en Li-O-overflade, som repræsenterer elektrodeoverfladen i et lithium-ion-batteri. DFT-beregningerne viste, at vandmolekyler danner stærke hydrogenbindinger med iltatomerne på overfladen, hvilket skaber et vandnetværk, der effektivt blokerer for migrationen af lithiumioner fra elektroden. Denne blokerende effekt er ansvarlig for passiveringen af elektrodeoverfladen og reduktionen af batteriets ydeevne over tid.
Samlet set giver den teoretiske ramme udviklet af forskerholdet en omfattende forståelse af vandmolekylers rolle i materialesyntese og fremstillingsprocesser, der involverer ioner. Resultaterne bidrager til det rationelle design og optimering af elektrokemiske reaktioner og batterisystemer, hvilket muliggør mere effektive og holdbare materialer og enheder.
 Varme artikler
Varme artikler
-
 Oligomerer observerede at efterligne kombinationen af DNA-strengeKredit:CC0 Public Domain Et internationalt forskerhold har for første gang observeret dynamiske kovalente oligomerer, der efterligner kombinationen af komplementære DNA-strenge, hvilket kan føre
Oligomerer observerede at efterligne kombinationen af DNA-strengeKredit:CC0 Public Domain Et internationalt forskerhold har for første gang observeret dynamiske kovalente oligomerer, der efterligner kombinationen af komplementære DNA-strenge, hvilket kan føre -
 Forskere udvikler mikrobobler til at ødelægge farlige biofilmProfessor i kemisk og biomolekylær ingeniørvidenskab Simon Rogers, venstre, postdoktorale forskere Jun Pong Park og Yongbeom Seo og professor i kemisk og biomolekylær teknik Hyunjoon Kong ledede et in
Forskere udvikler mikrobobler til at ødelægge farlige biofilmProfessor i kemisk og biomolekylær ingeniørvidenskab Simon Rogers, venstre, postdoktorale forskere Jun Pong Park og Yongbeom Seo og professor i kemisk og biomolekylær teknik Hyunjoon Kong ledede et in -
 En lettere receptKredit:Pixabay Type I-diabetespatienter injicerer typisk insulin flere gange om dagen, en smertefuld proces, der reducerer livskvaliteten. Injicerbare medicin er også forbundet med manglende overh
En lettere receptKredit:Pixabay Type I-diabetespatienter injicerer typisk insulin flere gange om dagen, en smertefuld proces, der reducerer livskvaliteten. Injicerbare medicin er også forbundet med manglende overh -
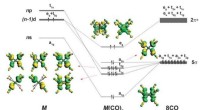 Kemikere viser, at 18-elektronprincippet ikke er begrænset til overgangsmetallerBindingsskema og form af de besatte valensorbitaler af M(CO) 8 (M =Ca, Sr, eller Ba). Opdeling af spd -valensorbitalerne i et atom M med konfigurationen (n - 1) d 2 ns 0 np 0 i oktakoordinatku
Kemikere viser, at 18-elektronprincippet ikke er begrænset til overgangsmetallerBindingsskema og form af de besatte valensorbitaler af M(CO) 8 (M =Ca, Sr, eller Ba). Opdeling af spd -valensorbitalerne i et atom M med konfigurationen (n - 1) d 2 ns 0 np 0 i oktakoordinatku
- En lysfølsom forbindelse muliggør varmefri membranmodulering i fotoswitches
- Kæmpe radiogalakse opdaget af astronomer
- La Niña kommer, hvilket øger chancerne for en farlig atlantisk orkansæson
- Hvordan Taiwan bruger buddhistisk litteratur til miljøundervisning
- Forskere:Vind, tørke forværre brande, ikke dårlig ledelse
- Nye fotoresponsive hydrogeler udviklet med øje for biomedicinske applikationer