


Løsning af enkeltmolekylmobilitet

Figur 1:Molekylære versioner af enheder som computerchips er nu et skridt tættere takket være en ny mikroskopistudie af forskere i Japan. Copyright:2010 iStockphoto/imagestock
Nanoteknologer samler indviklede nanodele, såsom computerchips, molekyle for molekyle ved hjælp af 'bottom-up' teknikker, der afspejler naturen. Én fremgangsmåde kører molekyler langs overflader til nye og funktionelle arrangementer ved hjælp af elektroner fra et scanningstunnelmikroskop (STM). Imidlertid, fordi energioverførsel mellem atomspidsen og overfladekemikaliet involverer mange komplekse interaktioner, møjsommelige anstrengelser er i øjeblikket nødvendige for at forstå selv de simpleste reaktioner.
Resultater fra et nyt teoretisk og eksperimentelt studie, imidlertid, kan snart give ikke-specialister mulighed for nemt at konstruere molekylære enheder. Kenta Motobayashi og Yousoo Kim fra RIKEN Advanced Science Institute i Wako og deres kolleger fra RIKEN og japanske universiteter har udviklet en matematisk formel, der beskriver, hvordan STM-inducerede molekylære vibrationer kobles sammen med dynamiske bevægelser på overflader – hvilket muliggør præcis beregning af energien og antallet af elektroner, der er nødvendige for at igangsætte enkelt molekylebevægelser.
Når videnskabsmænd bruger en STM til at udføre en ligetil molekylær bevægelse - f.eks. gør carbonmonoxid (CO) forbindelser til at 'hoppe' på palladiumoverflader - de ser, at brøkdelen af vellykkede bevægelser afhænger meget af den påførte spænding. For CO, dette er fordi at hoppe fra et overfladested til et andet kræver, at en tunnelelektron initierer en specifik strækningsvibration. I spændingsområdet svarende til denne vibrationsenergi, CO -hopping kan stige eksponentielt, giver anledning til såkaldte 'actionspektre':bevægelseskurver giver kontra spænding med former, der er karakteristiske for bestemte overfladereaktioner.
Motobayashi, Kim og kolleger forsøgte at afdække de mikroskopiske mekanismer bag STM-stimuleret diffusion ved at foreslå en formel, der relaterer bevægelsesudbytter til den energioverførselseffektivitet, der er nødvendig for at ophidse reaktionsudløsende vibrationer, samtidig med at der tages højde for termiske interaktioner. Montering af CO -actionspektre til denne formel afslørede de nøjagtige størrelser af kritiske reaktionsegenskaber, ligesom vibrationsenergier og hastighedskonstanter, fordi spektralkurverne var meget følsomme over for en lille ændring af tilpasningsparametrene.
Desuden, holdets nye ligning viste sig alsidig nok til at analysere de mere komplekse bevægelser af buten (C 4 H 8 ) molekyler på palladium, en proces, der involverer flere excitationer. Analyse af butenhandlingsspektrene med formlen viste tilstedeværelsen af tre forskellige vibrationer og muliggjorde beregning af reaktionsrækkefølgen - en grundlæggende kemisk egenskab, der identificerer antallet af tunnelelektroner, der er nødvendige for at starte overfladebevægelse.
Ifølge Motobayashi, de overraskende evner ved denne enkle metode bør udvide bottom-up nanoteknologipraksis. "STM-baseret aktionsspektroskopi, som præcist kan identificere kemiske arter takket være vores spektrale beslag, lover at bidrage meget til teknikken til sammensætning af molekylære enheder, ”Fastslår han.
Sidste artikelStøjen om grafen
Næste artikelGennembrud i vækst af nanokrystaller
 Varme artikler
Varme artikler
-
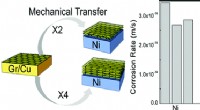 Graphen er den tyndeste kendte antikorrosionsbelægningNy forskning har etableret mirakelmaterialet kaldet grafen som verdens tyndeste kendte belægning til beskyttelse af metaller mod korrosion. Deres undersøgelse af denne potentielle nye brug af grafen v
Graphen er den tyndeste kendte antikorrosionsbelægningNy forskning har etableret mirakelmaterialet kaldet grafen som verdens tyndeste kendte belægning til beskyttelse af metaller mod korrosion. Deres undersøgelse af denne potentielle nye brug af grafen v -
 Elektron snigskytte mål grafenPå grund af dets spændende egenskaber kan grafen være det ideelle materiale til at bygge nye slags elektroniske enheder såsom sensorer, skærme, eller endda kvantecomputere. En af nøglerne til at udny
Elektron snigskytte mål grafenPå grund af dets spændende egenskaber kan grafen være det ideelle materiale til at bygge nye slags elektroniske enheder såsom sensorer, skærme, eller endda kvantecomputere. En af nøglerne til at udny -
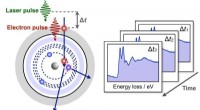 Ny teknik til at udforske strukturelle dynamikker i nanoworldKorte elektronpulser ophidser dybe elektroner på kerne-niveau i materialer, der giver øjebliksbilleder af den strukturelle dynamik efter laser-excitation. Kredit:Zewail Lab/Caltech En ny teknik ti
Ny teknik til at udforske strukturelle dynamikker i nanoworldKorte elektronpulser ophidser dybe elektroner på kerne-niveau i materialer, der giver øjebliksbilleder af den strukturelle dynamik efter laser-excitation. Kredit:Zewail Lab/Caltech En ny teknik ti -
 Kemikere udskriver sensorer til nanoobjekterKredit:CC0 Public Domain Unge forskere fra ITMO University foreslog en ny type optiske nano-sensorer. Deres funktionsprincip er baseret på lysets interaktion i tynde film:en lignende effekt kan ob
Kemikere udskriver sensorer til nanoobjekterKredit:CC0 Public Domain Unge forskere fra ITMO University foreslog en ny type optiske nano-sensorer. Deres funktionsprincip er baseret på lysets interaktion i tynde film:en lignende effekt kan ob
- Luftforurening dræber over 500, 000 europæere om året:rapport
- Indlejret dråbeprint-teknologi udskriver og behandler kontrollerbart dråber, der er suspenderet på…
- Verdens første digitale snevejledning
- Skal forældre hjælpe deres børn med lektier?
- Forskere kortlægger strukturen af det centrale kromatin-ombygningskompleks
- Forskere udfører kemisk undersøgelse af en gammel, metalrig kuglehob