


Et nyt blik på uordnet kulstof
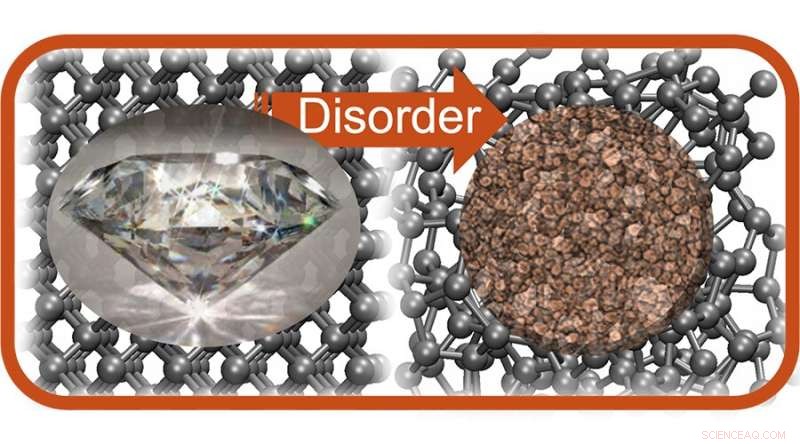
I modsætning til diamanter (venstre) og grafit har amorft kulstof (højre) ingen krystallinsk struktur; PME-forskere har nu kastet nyt lys over dets elektroniske egenskaber. Kredit:Galli Group
Når kulstofatomer stables i en perfekt gentagende tredimensionel krystal, kan de danne dyrebare diamanter. Arrangeret på en anden måde, i gentagne flade ark, laver carbon den skinnende grå grafit, der findes i blyanter. Men der er andre former for kulstof, der er mindre godt forstået. Amorft kulstof - normalt et sodet sort materiale - har ingen gentagen molekylær struktur, hvilket gør det udfordrende at studere.
Nu har forskere ved University of Chicagos Pritzker School of Molecular Engineering (PME) brugt en ny ramme til at forstå amorft kulstofs elektroniske egenskaber. Deres resultater lader videnskabsmænd bedre forudsige, hvordan materialet leder elektricitet og absorberer lys, og de blev offentliggjort i Proceedings of the National Academy of Sciences .
"Vi er nødt til at forstå, hvordan uordnet kulstof virker på et molekylært niveau for at kunne konstruere dette materiale til anvendelser som solenergikonvertering," sagde Giulia Galli, Liew-familieprofessor i molekylær teknik og professor i kemi ved University of Chicago. Galli har også en ansættelse som seniorforsker ved Argonne National Laboratory, hvor hun er direktør for MICCoM-centret.
I årtier har videnskabsmænd modelleret den måde, atomerne bevæger sig i amorft kulstof ved hjælp af lovene i klassisk mekanik – det sæt af ligninger, der for eksempel beskriver, hvordan en bil accelererer, eller hvordan en bold falder gennem luften. For nogle tunge atomer i det periodiske system er disse klassiske ligninger en god tilnærmelse til nøjagtigt at fange mange af materialernes egenskaber. Men for mange former for kulstof, og især amorfe kulstoffer, har holdet ledet af Galli fundet ud af, at det kommer til kort at bruge disse klassiske ligninger til at beskrive atomernes bevægelse.
"Amorft kulstof har mange egenskaber, der gør det værdifuldt til en række anvendelser, men det er udfordrende at modellere og simulere dets egenskaber på det grundlæggende niveau," sagde postdoktoralforsker Arpan Kundu, Ph.D., den første forfatter af papiret.
Galli har brugt de sidste tredive år på at udvikle og anvende kvantemekaniske metoder til at modellere og simulere egenskaberne af molekyler og faste stoffer. Hun undersøgte oprindeligt amorft kulstof helt i begyndelsen af sin karriere, og hun er for nylig vendt tilbage til udfordringen med ny indsigt.
Galli, Kundu og bachelorfysikforsker Yunxiang (Tony) Song udførte nye simuleringer af amorft kulstofs elektroniske egenskaber, denne gang ved at integrere kvanteprincipper til at beskrive bevægelserne af både elektroner og kerner i kulstofatomer. De fandt ud af, at det er afgørende at bruge kvantemekanik for begge - i stedet for klassisk mekanik for kernerne - for nøjagtigt at forudsige egenskaberne af amorft kulstof.
For eksempel ved at bruge deres raffinerede, kvantemekaniske modeller, forudsagde PME-teamet en højere elektrisk ledningsevne, end man ellers ville have forventet.
Resultaterne rapporteret i PNAS artiklen er nyttige ikke kun til at forstå amorft kulstof, men også andre lignende amorfe faste stoffer, sagde forskerne. Men de påpegede også, at der er meget mere arbejde at gøre - uordnede kulstofmaterialer kan udvise radikalt forskellige egenskaber afhængigt af deres tæthed, hvilket igen afhænger af den metode, der bruges til at fremstille materialet.
"Når noget er arrangeret i en krystal, ved du præcis, hvad dets struktur er, men når det først er uordnet, kan det være uordnet på mange mulige måder," sagde Kundu.
Holdet planlægger at fortsætte med at studere amorft kulstof og dets potentielle anvendelser. + Udforsk yderligere
Oprindelse af bosontoppen i amorfe faste stoffer
 Varme artikler
Varme artikler
-
 Kemikere opnår gennembrud i syntesen af grafen nanobåndKredit:CC0 Public Domain Grafen nanobånd kan snart blive meget nemmere at producere. Et internationalt forskerhold ledet af Martin Luther University Halle-Wittenberg (MLU), University of Tennessee
Kemikere opnår gennembrud i syntesen af grafen nanobåndKredit:CC0 Public Domain Grafen nanobånd kan snart blive meget nemmere at producere. Et internationalt forskerhold ledet af Martin Luther University Halle-Wittenberg (MLU), University of Tennessee -
 Banebrydende fund om nanopartikellevering af HIV/AIDS-medicin til hjernenKredit:CC0 Public Domain En biokemiforsker fra University of Miami Miller School of Medicine har fundet ud af, at et nanopartikel-lægemiddelleveringssystem kan reducere HIV/AIDS-virale reservoirer
Banebrydende fund om nanopartikellevering af HIV/AIDS-medicin til hjernenKredit:CC0 Public Domain En biokemiforsker fra University of Miami Miller School of Medicine har fundet ud af, at et nanopartikel-lægemiddelleveringssystem kan reducere HIV/AIDS-virale reservoirer -
 Guld nanopartikler, der får blade til at lyse i mørketBilledkredit: Nanoskala , DOI:10.1039/C0NR00330A. For flere detaljer, se den originale publikation. (PhysOrg.com)-Forskere i Taiwan tror, at de i sidste ende muligvis kan erstatte gadelamper me
Guld nanopartikler, der får blade til at lyse i mørketBilledkredit: Nanoskala , DOI:10.1039/C0NR00330A. For flere detaljer, se den originale publikation. (PhysOrg.com)-Forskere i Taiwan tror, at de i sidste ende muligvis kan erstatte gadelamper me -
 Ny enhed kan gøre konvertering af spildvarme til elektricitet industrielt konkurrencedygtigDen foreslåede termoelektriske enhed består af mange parallelle nanotråde med en ekstern portspænding, der kan indstilles for at optimere effektiviteten og effektudbyttet til forskellige temperaturfor
Ny enhed kan gøre konvertering af spildvarme til elektricitet industrielt konkurrencedygtigDen foreslåede termoelektriske enhed består af mange parallelle nanotråde med en ekstern portspænding, der kan indstilles for at optimere effektiviteten og effektudbyttet til forskellige temperaturfor
- Hvad er mycelia i mikrobiologi?
- Forskere demonstrerer første terahertz kvantesansning
- Den usikre enhjulede cykel, der lærte sig selv, og hvordan den hjælper AI med at træffe gode besl…
- Tager temperaturen af mørkt stof
- Topmoderne lasere på mikroniveau
- Majsproduktivitet i realtid:Satellitter, feltkameraer, og landmænd går sammen