


Maskinlæring vejleder kulstofnanoteknologi
Carbon nanostrukturer kan blive nemmere at designe og syntetisere takket være en maskinlæringsmetode, der forudsiger, hvordan de vokser på metaloverflader. Den nye tilgang, udviklet af forskere ved Japans Tohoku University og Kinas Shanghai Jiao Tong University, vil gøre det lettere at udnytte den unikke kemiske alsidighed af kulstofnanoteknologi. Metoden blev publiceret i tidsskriftet Nature Communications .
Væksten af kulstofnanostrukturer på en række forskellige overflader, herunder som atomisk tynde film, er blevet undersøgt i vid udstrækning, men lidt er kendt om dynamikken og atomniveaufaktorerne, der styrer kvaliteten af de resulterende materialer. "Vores arbejde adresserer en afgørende udfordring for at realisere potentialet af kulstof-nanostrukturer i elektronik- eller energibehandlingsenheder," siger Hao Li fra Tohoku University-teamet.
Det brede udvalg af mulige overflader og processens følsomhed over for flere variabler gør direkte eksperimentel undersøgelse udfordrende. Forskerne henvendte sig derfor til maskinlæringssimuleringer som en mere effektiv måde at udforske disse systemer på.
Med maskinlæring kan forskellige teoretiske modeller kombineres med data fra kemieksperimenter for at forudsige dynamikken i kulstofkrystallinsk vækst og bestemme, hvordan den kan kontrolleres for at opnå specifikke resultater. Simuleringsprogrammet udforsker strategier og identificerer, hvilke der virker, og hvilke der ikke gør, uden at det er nødvendigt for mennesker at guide hvert trin i processen.
-
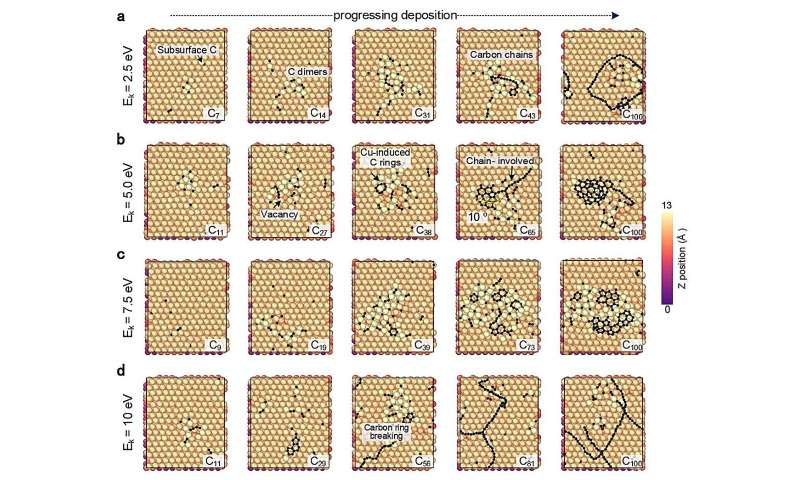
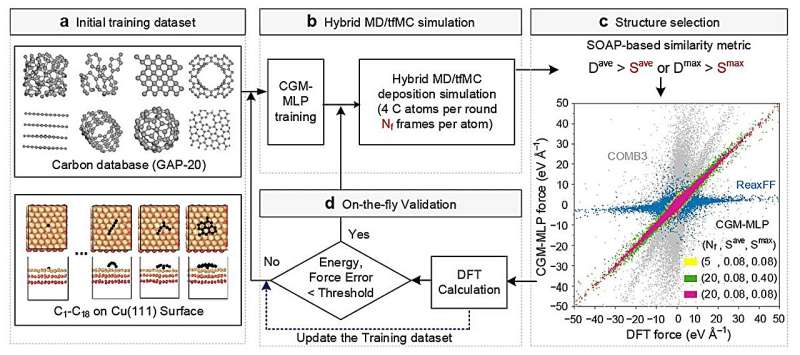
CGM-MLP-drevne simuleringer af grafenvækst på Cu(111) med forskellige kulstofindfaldende kinetiske energier (Ek). (a) 2,5 eV, (b) 5,0 eV, (c) 7,5 eV og (d) 10 eV. Kredit:Nature Communications (2024). DOI:10.1038/s41467-023-44525-z -
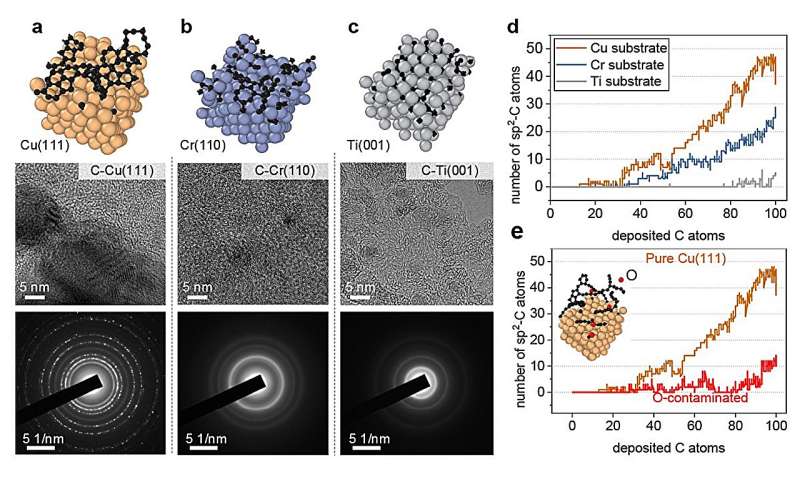
Repræsentative metalliske overflader til vækst af kulstofnanostrukturer. (a) ren Cu(111), (b) Cr(110 og (c) Ti(001) overflade. Under hver overflade, højopløsningstransmissionselektronmikroskopi (HRTEM) billeder og udvalgt område elektrondiffraktion (SAED) billeder af kulstof nanostrukturer fremstillet ved magnetronforstøvningsaflejring er tilvejebragt (d) Antallet af sp 2 -C som funktion af aflejrede carbonatomer på forskellige metalsubstrater og e O-forurenet Cu(111). Kredit:Nature Communications (2024). DOI:10.1038/s41467-023-44525-z
Forskerne testede denne tilgang ved at undersøge simuleringer af væksten af grafen, en form for kulstof, på en kobberoverflade. Efter at have etableret den grundlæggende ramme viste de, hvordan deres tilgang også kunne anvendes på andre metaloverflader, såsom titanium, krom og kobber forurenet med oxygen.
Fordelingen af elektroner omkring kernerne af atomer i forskellige former for grafenkrystaller kan variere. Disse subtile forskelle i atomstruktur og elektronarrangement påvirker materialets overordnede kemiske og elektrokemiske egenskaber. Maskinlæringstilgangen kan teste, hvordan disse forskelle påvirker diffusionen af individuelle atomer og bundne atomer og dannelsen af kulstofkæder, buer og ringstrukturer.
Holdet validerede resultaterne af simuleringerne gennem eksperimenter og fandt ud af, at de passede tæt sammen. "Samlet set giver vores arbejde en praktisk og effektiv metode til at designe metal- eller legeringssubstrater for at opnå ønskede kulstofnanostrukturer og udforske yderligere muligheder," siger Li.
Han tilføjer, at det fremtidige arbejde vil bygge videre på dette for at undersøge emner som grænseflader mellem faste stoffer og væsker i avancerede katalysatorer og de kemiske egenskaber af materialer, der anvendes til behandling og lagring af energi.
Flere oplysninger: Di Zhang et al., Aktiv maskinlæringsmodel for dynamiske simulerings- og vækstmekanismer af kulstof på metaloverfladen, Nature Communications (2024). DOI:10.1038/s41467-023-44525-z
Leveret af Tohoku University
Sidste artikelForskere udvikler antiviral farve nanocoating-teknologi
Næste artikelSådan gør du lyse kvanteprikker endnu lysere
 Varme artikler
Varme artikler
-
 Kulstof i farve:Først nogensinde farvede tynde film af nanorør oprettetPrøver af de farverige tynde film af carbon nanorør, som fremstillet i fremstillingsreaktoren. Kredit:Aalto University En metode udviklet ved Aalto University, Finland, kan producere store mængder
Kulstof i farve:Først nogensinde farvede tynde film af nanorør oprettetPrøver af de farverige tynde film af carbon nanorør, som fremstillet i fremstillingsreaktoren. Kredit:Aalto University En metode udviklet ved Aalto University, Finland, kan producere store mængder -
 Mikrostrukturer samles selv til nye materialerIngeniører dyrkede terninger af nanoarkitekteret materiale for at teste dets styrke og modstandsdygtighed. Kredit:Greer Lab/Caltech En ny proces udviklet hos Caltech gør det for første gang muligt
Mikrostrukturer samles selv til nye materialerIngeniører dyrkede terninger af nanoarkitekteret materiale for at teste dets styrke og modstandsdygtighed. Kredit:Greer Lab/Caltech En ny proces udviklet hos Caltech gør det for første gang muligt -
 Forbedrede superkondensatorer til superbatterier, elektriske køretøjer(a) Dette er en skematisk illustration af fremstillingsprocessen for RGM nanostrukturskum. SEM-billeder af (b–c) as-grown GM-skum (d) Let belastet RGM, og (e) tungt belastet RGM. Kredit:UC Rivers
Forbedrede superkondensatorer til superbatterier, elektriske køretøjer(a) Dette er en skematisk illustration af fremstillingsprocessen for RGM nanostrukturskum. SEM-billeder af (b–c) as-grown GM-skum (d) Let belastet RGM, og (e) tungt belastet RGM. Kredit:UC Rivers -
 Nye nanopartikeloverbygninger lavet af pyramideformede byggeklodserI forskning, der kan hjælpe med at bygge bro mellem nano og makro, Brown University kemikere har brugt pyramideformede nanopartikler til at skabe, hvad der kan være den mest komplekse makroskala overb
Nye nanopartikeloverbygninger lavet af pyramideformede byggeklodserI forskning, der kan hjælpe med at bygge bro mellem nano og makro, Brown University kemikere har brugt pyramideformede nanopartikler til at skabe, hvad der kan være den mest komplekse makroskala overb
- Primordiale sorte huller og søgen efter mørkt stof fra multiverset
- Åh de GAN'er:Scanner-fingerteknik kan resultere i falske fingeraftryk
- Sådan opretter du et 3D vådområde Diorama
- Mekanisk risproduktion
- One night brand:Sexede snaps fører til rene køb
- Kæmpe synkehul truer med at sluge det mexicanske hjem