


Bioinformatikere slipper af med et unødvendigt trin i proteinstabilitetsanalyse
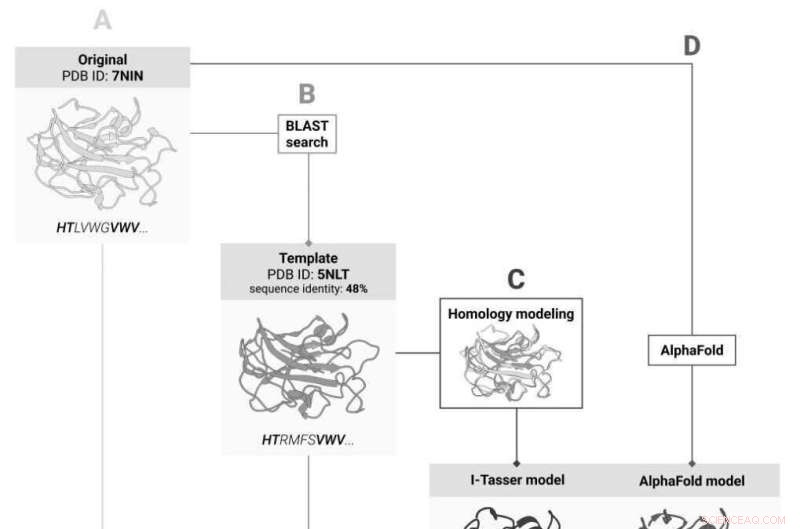
Fire måder at forudsige ændringer i proteinstabilitet efter mutation:(A) ved strukturen af det oprindelige protein; (B) ved strukturen af dets homolog; (C) af strukturen af det oprindelige protein forudsagt baseret på strukturen af holomlogen, og (D) af strukturen forudsagt af kunstig intelligens baseret på aminosyresekvensen. Kredit:Skolkovo Institut for Videnskab og Teknologi
Forskere fra Skoltech Center for Molekylær og Cellulær Biologi sammenlignede forskellige metoder til forudsigelse af proteinstruktur med hensyn til evaluering af mutantproteinstabilitet og opnåede det samme resultat for de AI-forudsagte strukturer og eksperimentelle tredimensionelle (3D) af proteiner med lignende aminosyresekvenser. Forsøget på at forudsige det målrettede proteins struktur ud fra den kendte struktur af dets "slægtning" gjorde kun forudsigelsen mindre nøjagtig. Holdets resultater vil lette foreløbige beregninger i evalueringen af stabilitetsændringer forårsaget af mutation. Forskningen blev offentliggjort i Bioinformatics .
Biologiske eksperimenter involverer ofte mutante proteiner, som er nødvendige for studiet af proteinets struktur og funktioner eller celleprocesser, såvel som proteinteknologi. Mutationer vides at påvirke et proteins struktur og stabilitet. Da eksperimenter er for dyre og tidskrævende, skaber forskerne en løsning i form af beregningsmetoder til at evaluere mutationers indvirkning på stabiliteten. Deres anvendelser kræver dog viden om et proteins 3D-struktur.
En eksperimentel 3D-struktur er ikke tilgængelig for alle proteiner og mangler sandsynligvis for den, holdet er målrettet mod. Hvis dette er tilfældet, kan 3D-modeller af proteinets homologer, altså dets "nærmeste slægtninge", give livline, fordi graden af lighed i aminosyresekvenser, der sikrer et godt match mellem proteinernes 3D-strukturer, er velkendt. Løsningen ville være først at forudsige proteinets struktur baseret på den kendte struktur af dets homolog og derefter beregne virkningen af mutationer for den forudsagte model.
Takket være sidste års gennembrud inden for forudsigelse af proteinstruktur, har forskerne nu et alternativ:i stedet for at forudsige 3D-strukturen baseret på homologer, kan de bruge det AI-baserede AlphaFold-værktøj, som forudsiger proteinstrukturen ud fra aminosyresekvensen og allerede har beskæftiget sig med med langt de fleste proteiner kendt til dato.
I deres nylige undersøgelse besluttede Skoltech-forskerne at finde ud af, hvilken af disse tilgange der fungerer bedst til at forudsige stabilitetsændringer ved mutation. Hvor præcis AlphaFold end måtte være, er det stadig "guldstandarden" at finde proteinstrukturen gennem eksperimenter. Når de sammenlignede de to tilgange, brugte holdet syv stabilitetsevalueringsmetoder og sammenlignede deres resultater med resultaterne fra AlphaFold og I-Tasser, det bedste homologbaserede strukturforudsigelsessystem. Forskerne undersøgte også, om de kan springe den homolog-baserede strukturforudsigelse over og beregne stabilitet for den kendte struktur af det homologe protein.
"Vi besluttede at finde ud af, hvor langt vi ville afvige fra præcis forudsigelse, hvis vi brugte den 'naboende' proteinstruktur i stedet for den rigtige. Det viste sig, at det homologi-baserede forudsigelsestrin kun gør tingene værre ved at give et mindre præcist resultat. Vi har vist, at det stort set ikke gør nogen forskel, om du bruger homologens eksperimentelle struktur eller AlphaFolds forudsigelse. På en måde handlede det om validering:Når du står over for en ny metode, skal du tjekke, om den fungerer til din opgave i første omgang Det er præcis, hvad vi gjorde," første forfatter til undersøgelsen, Skoltech Ph.D. studerende Marina Pak, kommentarer.
"Med al denne ballade over AlphaFold mener nogle forskere og ikke-professionelle, at det har løst alle proteinforskningsspørgsmål inden for beregningsbiologi, men det har det ikke. For eksempel viser forudsigelsen af mutationsinducerede stabilitetsændringer stadig ret lav pålidelighed, selv selvom ændringen i stabilitet er blandt de vigtigste drivkræfter bag proteinfunktionalitet. Et værktøj, der utvetydigt kunne bestemme mutationens indvirkning på stabiliteten, ville hjælpe både med at planlægge eksperimentet og fortolke resultaterne. Antag, at for et protein, der ikke er optimalt mht. stabilitet, ønsker vi at finde mutationer, der vil gøre det stabilt under de ønskede forhold, for eksempel sikre, at det forbliver aktivt ved høj temperatur. Når vi kan gøre dette gennem beregninger alene, vil tilgangen til protein redesign og optimering ændre sig dramatisk." hovedforfatter af undersøgelsen, konkluderer Skoltech-assistentprofessor Dmitry Ivankov.
Selvom forudsigelse af stabilitetsændringer ser ud til at være nemmere end at forudsige 3D-strukturen, er det stadig en vanskelig udfordring selv for kunstig intelligens. Knappe træningsdata er kun et af problemerne:AlphaFold havde næsten 200.000 proteinstrukturer at træne, men eksperimentelle data om stabilitetsændringer beløber sig til tusindvis af sæt, mens de kun dækkede et par dusin unikke proteiner. Forfatterne håber, at hvis flere data bliver tilgængelige, og forskerne viser en større interesse for opgaven, vil et gennembrud med sikkerhed snart ske. + Udforsk yderligere
Fysikere bruger kunstig intelligens til at finde de hidtil mest komplekse proteinknuder
 Varme artikler
Varme artikler
-
 Mere komplekse biologiske systemer udvikler sig mere fritFørste forfatter Mato Lagator analyserer fænotypen af en prøve af E coli mutanter. Kredit:IST Østrig Vores gener (alias genotypen) bestemmer vores karakteristika (aka. fænotypen). Evolution vi
Mere komplekse biologiske systemer udvikler sig mere fritFørste forfatter Mato Lagator analyserer fænotypen af en prøve af E coli mutanter. Kredit:IST Østrig Vores gener (alias genotypen) bestemmer vores karakteristika (aka. fænotypen). Evolution vi -
 Forskere finder, at der er mindst 14, 003 plantetyper i Amazonas-bassinetUkontaktet indfødt stamme i den brasilianske delstat Acre. Kredit:Gleilson Miranda / Governo do Acre / Wikipedia (Phys.org) – Et stort team af forskere fra Brasilien, England., Columbia og Spanien
Forskere finder, at der er mindst 14, 003 plantetyper i Amazonas-bassinetUkontaktet indfødt stamme i den brasilianske delstat Acre. Kredit:Gleilson Miranda / Governo do Acre / Wikipedia (Phys.org) – Et stort team af forskere fra Brasilien, England., Columbia og Spanien -
 Translokerede høge trives i HispaniolaTranslocated Ridgways Hawks trives i deres nye hjem på ejendommen til et feriested på øen Hispaniola. Kredit:Russell Thorstrom Arttranslokation - at fange dyr på ét sted og frigive dem på et andet
Translokerede høge trives i HispaniolaTranslocated Ridgways Hawks trives i deres nye hjem på ejendommen til et feriested på øen Hispaniola. Kredit:Russell Thorstrom Arttranslokation - at fange dyr på ét sted og frigive dem på et andet -
 Kofugleunger har det bedst med to sangfugle-redekammerater – ikke fire, ikke nul, undersøgelsesfundEn kofugl, der sætter sig ned, kigger ud fra en sangered. Kredit:Nicholas Antonson Brunhovedet kofugle er generalistiske yngleparasitter, der lægger deres æg i rederne hos mange andre fuglearter og
Kofugleunger har det bedst med to sangfugle-redekammerater – ikke fire, ikke nul, undersøgelsesfundEn kofugl, der sætter sig ned, kigger ud fra en sangered. Kredit:Nicholas Antonson Brunhovedet kofugle er generalistiske yngleparasitter, der lægger deres æg i rederne hos mange andre fuglearter og
- De mest markante ændringer i britisk luftkvalitet under lockdown har været i byområder, anmeldels…
- Hvilke nye emojis kommer?
- Transportsystemet for planter og dyr
- Dovenskab førte til udryddelse af Homo erectus
- Studiebuster 9 til 5 model til akademisk arbejde
- Scotch tippede til at slå sprutrekord på $1 mio. plus