


Sådan finder du markørgener i celleklynger
Grafisk abstrakt. Kredit:Journal of Molecular Biology (2022). DOI:10.1016/j.jmb.2022.167525
Hvilke gener er specifikke for en bestemt celletype, dvs. "mærker" deres identitet? Med den stigende størrelse af datasæt i dag, er det ofte en udfordring at besvare dette spørgsmål. Ofte er markørgener simpelthen gener, der er blevet fundet i specifikke cellepopulationer. Imidlertid kunne mange flere gener være karakteristiske for en bestemt celletype, men forblive uopdagede.
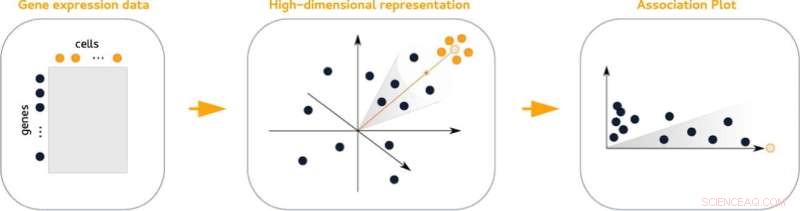
Association plots (APL), en ny statistisk metode til at visualisere genaktivitet i en celleklynge, gør det lettere at finde dets markørgener. Plottene sammenligner aktiviteten af gener fra en given klynge med alle andre klynger fra datasættet. Derudover gør de det nemt at se, hvilke gener der deles med andre klynger.
"Associationsplot giver os ikke kun mulighed for at identificere nye markørgener. Det virker også omvendt - vi er i stand til at matche klynger af ukendt identitet i et datasæt til celletyper baseret på en medfølgende liste over markørgener," siger Elzbieta Gralinska fra Max Planck Institute for Molecular Genetics i Berlin.
Bioteknologen arbejder i teamet af Martin Vingron, som udviklede teknikken. Forskerne demonstrerede teknikkens funktionalitet på to offentligt tilgængelige datasæt og offentliggjorde resultaterne i Journal of Molecular Biology . Desuden er APL blevet frigivet som et gratis modul til det statistiske miljø R. APL-pakken giver forskere mulighed for visuelt at inspicere deres enkeltcelledata og vælge individuelle gener med markøren for at lære mere dybdegående detaljer.
Analyse og gruppering af enkeltceller
Hvorfor er det nødvendigt at identificere markørgener i første omgang? Moderne sekventeringsteknologier er i stand til at dechifrere individuelle RNA-molekyler i individuelle celler. Fra en blodprøve kan for eksempel hver celle adskilles, og en prøve af cellens RNA'er kan afkodes. Disse enkeltcelledata repræsenterer de aktive gener, der blev transskriberet til RNA-molekyler.
Fordelen:I stedet for at gå i tvivl om, hvilken celletype et bestemt RNA tilhører, kan det spores tilbage til dets oprindelsescelle. Ulempen:sekventering af tusindvis af RNA'er i hver enkelt celle ud af titusindvis af celler producerer ekstraordinære mængder data.
En udvej er at sortere cellerne ud fra deres RNA-indhold. "Enkeltcelledata er sammensat af en vild blanding af mange forskellige celletyper. Vi er interesserede i celler af samme celletype, som alle burde opføre sig ens," forklarer Martin Vingron. Derfor giver det mening at gruppere lignende celler beregningsmæssigt, siger han. "For os definerer markørgenerne en celletype."
Udforsker celleklynger interaktivt
Ved hjælp af offentligt tilgængelige data fra hvide blodlegemer demonstrerede holdet, hvordan den nye algoritme virker. De mange forskellige typer hvide blodlegemer som T-celler, B-celler eller monocytter er alle grupperet i separate klynger. Forskerne bekræftede kendte markørgener og var i stand til at vise, at nære slægtninge blandt blodcellerne også deler stor lighed i deres genaktivitet.
"Hvert af de markørgener, vi fandt med APL, kunne være blevet opdaget af mindst én anden eksisterende metode til identifikation af markørgener," siger Gralinska. Men fordelen ved APL i forhold til de eksisterende algoritmer er dens grafiske repræsentation af resultaterne, siger hun. "Eksisterende værktøjer giver lange lister over gener og scoreværdier. Ofte går brugerne igennem listen og stopper ved en vilkårlig cut-off."
I modsætning hertil giver den nye metode en måde at visualisere disse gener, klikke på hver enkelt og se nærmere på dens aktivitet, siger hun. "Vi leverer ikke kun lister over markørgener, vi giver brugerne mulighed for at gennemgå, hvordan disse gener opfører sig," siger forskeren. "Med associationsplot kan de dykke ned i deres data for at lære mere om hver celletype." Derudover, siger hun, er det meget nemt at nedbryde de mest interessante geners biologiske rolle i et efterfølgende trin via Gene Ontology termberigelsesanalyse, som er kompatibel med APL-softwaren – noget hun betragter som "en meget nyttig funktion."
Den underliggende matematiske model
De højdimensionelle data, der indeholder information om aktivitet på tværs af gener, kan ikke repræsenteres visuelt uden tab af information. Det samme gælder for klyngede data, som alle komplicerer analyse. "Vores trick er, at vi tager højde for mange flere end blot to eller tre dimensioner, men i sidste ende skaber et todimensionelt diagram," siger Gralinska.
Associationsplottene er afledt af en matematisk teknik, der samtidigt indlejrer både gener og celler i et fælles højdimensionelt rum. Måling af afstandene mellem gener og en given celleklynge i dette rum resulterer i værdipar, der afspejler associationen af et gen til en given klynge og giver indsigt i dets association til andre klynger.
"En mangel ved APL er, at vi er afhængige af præ-klyngede data, hvilket betyder, at vi er nødt til at stole på andre teknikker til klyngedannelse," siger Martin Vingron. "Alligevel håber vi, at vores nye metode vil finde mange nye brugere. Vi oplever, at en visuel og interaktiv proces simpelthen giver en bedre analyse."
 Varme artikler
Varme artikler
-
 Svømmerkløe:Hvad forårsager denne oversete sneglebårne sygdom?Feltstudie i udbrudsområdet. Kredit:Professor Dr. Duangduen Krailas Cercarial dermatitis, også kendt som svømmekløe eller muslingegraverkløe, er forårsaget af larverne af blodslynge, der er parasit
Svømmerkløe:Hvad forårsager denne oversete sneglebårne sygdom?Feltstudie i udbrudsområdet. Kredit:Professor Dr. Duangduen Krailas Cercarial dermatitis, også kendt som svømmekløe eller muslingegraverkløe, er forårsaget af larverne af blodslynge, der er parasit -
 Nordamerika først:Forskere offentliggør videnskabelig undersøgelse om cannabisproduktionCannabis indica. Kredit:Wikipedia University of Guelph forskere har offentliggjort, hvad der menes at være den første videnskabelige artikel i Nordamerika om forbedring af medicinsk cannabisplante
Nordamerika først:Forskere offentliggør videnskabelig undersøgelse om cannabisproduktionCannabis indica. Kredit:Wikipedia University of Guelph forskere har offentliggjort, hvad der menes at være den første videnskabelige artikel i Nordamerika om forbedring af medicinsk cannabisplante -
 Hvordan fuglene og bierne hjælper kaffeplanterKredit:Shutterstock At nippe til en kop kaffe på vej til arbejde er et ritual, de fleste tager for givet uden at tænke på, hvordan de lækre kaffebønner nåede deres kop. Du ved sikkert, at det komme
Hvordan fuglene og bierne hjælper kaffeplanterKredit:Shutterstock At nippe til en kop kaffe på vej til arbejde er et ritual, de fleste tager for givet uden at tænke på, hvordan de lækre kaffebønner nåede deres kop. Du ved sikkert, at det komme -
 Skinde lys på viras sociale livVira som disse H1N1 -influenzapartikler er afhængige af værtsceller til at reproducere, men de kan stadig vise social adfærd, interagere med andre vira ved at konkurrere, samarbejde og nogle gange sny
Skinde lys på viras sociale livVira som disse H1N1 -influenzapartikler er afhængige af værtsceller til at reproducere, men de kan stadig vise social adfærd, interagere med andre vira ved at konkurrere, samarbejde og nogle gange sny
- Forskning afslører første bevis på RNA-udløst faseadskillelse
- Ruslands Nauka videnskabsmodul lægger til kaj med ISS
- Fornyelige, ikke-genanvendelige og uudtømmelige ressourcer
- På vej mod et nyt vandparadigme
- Kigger på atomare strukturer med ikke mere end blyant og papir
- Mantis-rejeklubbens mangefacetterede design er inspirerende avancerede kompositmaterialer til fly, f…