


Software -rammer designet til at fremskynde opdagelse af lægemidler vinder IEEE International Scalable Computing Challenge

Shantenu Jha, formand for Brookhaven Labs Center for Datadrevet Opdagelse, og hans team fra Rutgers University og University College London designet en software ramme til præcist og hurtigt at beregne, hvor stærkt lægemiddelkandidater binder til deres målproteiner. Rammerne er rettet mod at løse det virkelige problem med lægemiddeldesign-i øjeblikket en lang og dyr proces-og kan have indflydelse på personlig medicin. Kredit:Brookhaven National Laboratory
Løsninger på mange videnskabelige og tekniske problemer i den virkelige verden-fra at forbedre vejrmodeller og designe nye energimaterialer til at forstå, hvordan universet dannede sig-kræver applikationer, der kan skaleres til en meget stor størrelse og høj ydeevne. Hvert år, gennem sin International Scalable Computing Challenge (SCALE), Institute of Electrical and Electronics Engineers (IEEE) anerkender et projekt, der fremmer applikationsudvikling og understøtter infrastruktur for at muliggøre storstilet, højtydende computing nødvendig for at løse sådanne problemer.
Årets vinder, "Muliggør afvejning mellem nøjagtighed og beregningsomkostninger:adaptive algoritmer for at reducere tid til klinisk indsigt, "er resultatet af et samarbejde mellem kemikere og beregnings- og computerforskere ved det amerikanske energiministerium (DOE) Brookhaven National Laboratory, Rutgers University, og University College London. Teammedlemmerne blev hædret på det 18. IEEE/Association for Computing Machinery (ACM) International Symposium on Cluster, Cloud and Grid Computing afholdes i Washington, DC, fra 1. til 4. maj.
"Vi udviklede en numerisk beregningsmetode til nøjagtigt og hurtigt at evaluere effekten af forskellige lægemiddelkandidater, "sagde teammedlem Shantenu Jha, formand for Center for Datadrevet Opdagelse, del af Brookhaven Labs Computational Science Initiative. "Selvom vi endnu ikke har anvendt denne metode til at designe et nyt lægemiddel, vi demonstrerede, at det kunne fungere i de store skalaer, der er involveret i processen med opdagelse af lægemidler. "
Lægemiddelopdagelse er ligesom at designe en nøgle, der passer til en lås. For at et lægemiddel skal være effektivt til behandling af en bestemt sygdom, den skal binde tæt til et molekyle - normalt et protein - der er forbundet med denne sygdom. Først da kan lægemidlet aktivere eller hæmme funktionen af målmolekylet. Forskere kan screene 10, 000 eller flere molekylære forbindelser, inden man finder nogen, der har den ønskede biologiske aktivitet. Men disse "bly" -forbindelser mangler ofte styrken, selektivitet, eller stabilitet nødvendig for at blive et lægemiddel. Ved at ændre den kemiske struktur af disse kundeemner, forskere kan designe forbindelser med de passende lægemiddellignende egenskaber. De designede lægemiddelkandidater bevæger sig derefter langs udviklingsrørledningen til det prækliniske teststadium. Af disse kandidater, kun en lille brøkdel går ind i den kliniske forsøgsfase, og kun en ender med at blive et godkendt lægemiddel til patientbrug. At bringe et nyt lægemiddel på markedet kan tage et årti eller længere og koste milliarder af dollars.
Overvindelse af flaskehalse for lægemiddeldesign gennem beregningsvidenskab
Nylige fremskridt inden for teknologi og viden har resulteret i en ny æra med opdagelse af lægemidler - en der kan reducere tid og omkostninger betydeligt til lægemiddeludviklingsprocessen. Forbedringer i vores forståelse af biologiske molekylers 3D-krystalstrukturer og stigninger i computerkraft gør det muligt at bruge beregningsmetoder til at forudsige lægemiddel-målinteraktioner.
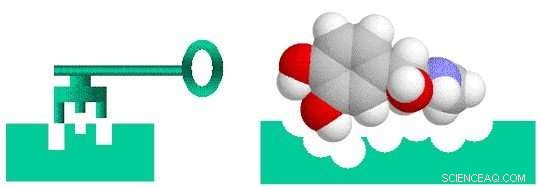
Lægemiddelopdagelse er et lås-og-nøgle-problem, hvor stoffet (nøglen) specifikt skal passe til det biologiske mål (lås). Kredit:Brookhaven National Laboratory
I særdeleshed, en computersimuleringsteknik kaldet molekylær dynamik har vist løfte om nøjagtigt at forudsige den styrke, hvormed lægemiddelmolekyler binder til deres mål (bindingsaffinitet). Molekylær dynamik simulerer, hvordan atomer og molekyler bevæger sig, når de interagerer i deres miljø. I tilfælde af stofopdagelse, simuleringerne afslører, hvordan lægemiddelmolekyler interagerer med deres målprotein og ændrer proteinets konformation, eller form, som bestemmer dens funktion.
Imidlertid, disse forudsigelsesegenskaber fungerer endnu ikke i stor nok eller hurtigt nok til at farmaceutiske virksomheder kan tage dem i deres udviklingsproces.
"At oversætte disse fremskridt inden for forudsigelig nøjagtighed til at påvirke industriel beslutningstagning kræver, at i størrelsesordenen 10, 000 bindingsaffiniteter beregnes så hurtigt som muligt, uden tab af nøjagtighed, "sagde Jha." At producere rettidig indsigt kræver en beregningseffektivitet, der er baseret på udviklingen af nye algoritmer og skalerbare softwaresystemer, og smart tildeling af supercomputingressourcer. "
Jha og hans samarbejdspartnere ved Rutgers University, hvor han også er professor i afdelingen for el- og computerteknik, og University College London designet en software -ramme, der understøtter præcis og hurtig beregning af bindende affiniteter, samtidig med at brugen af beregningsressourcer optimeres. Denne ramme, kaldet High-Throughput Binding Affinity Calculator (HTBAC), bygger på RADICAL-Cybertools-projektet, som Jha leder som hovedforsker af Rutgers 'Research in Advanced Distributed Cyberinfrastructure and Applications Laboratory (RADICAL). Målet med RADICAL-Cybertools er at levere en pakke software-byggesten til at understøtte arbejdsgange i store videnskabelige applikationer på højtydende computingplatforme, som samler computerkraft til at løse store beregningsproblemer, der ellers ville være uløselige på grund af den nødvendige tid.
Inden for datalogi, arbejdsgange refererer til en række behandlingstrin, der er nødvendige for at fuldføre en opgave eller løse et problem. Især for videnskabelige arbejdsgange, det er vigtigt, at arbejdsgangene er fleksible, så de dynamisk kan tilpasse sig under runtime for at give de mest nøjagtige resultater, mens de effektivt udnytter den tilgængelige computetid. Sådanne adaptive arbejdsgange er ideelle til opdagelse af lægemidler, fordi kun lægemidler med høje bindingsaffiniteter bør evalueres yderligere.
"Den ønskede afvejning mellem den krævede nøjagtighed og beregningsomkostninger (tid) ændres under hele lægemidlets opdagelse, når processen bevæger sig fra screening til valg af kundeemner og derefter blyoptimering, "sagde Jha." Et betydeligt antal forbindelser skal screenes billigt for at fjerne dårlige bindemidler, før der er behov for mere præcise metoder til at skelne mellem de bedste bindemidler. At levere den hurtigste tid til løsning kræver overvågning af simuleringernes forløb og beslutninger om fortsat udførelse baseret på videnskabelig betydning. "
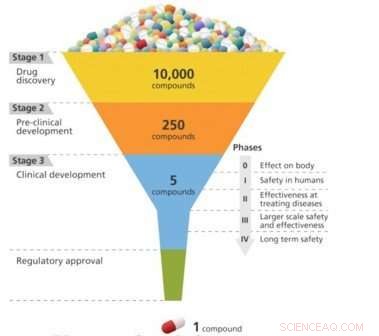
En skematisk oversigt over lægemiddeludviklingsprocessen, der gradvist finpudser de mest effektive kandidater fra en stor indledende pulje. Kredit:Brookhaven National Laboratory
Med andre ord, det ville ikke være fornuftigt at fortsætte simuleringer af en bestemt lægemiddel-protein-interaktion, hvis lægemidlet svagt binder proteinet i forhold til de andre kandidater. Men det ville være fornuftigt at tildele yderligere beregningsressourcer, hvis et lægemiddel viser en høj bindende affinitet.
Understøttelse af adaptive arbejdsgange i de store skalaer, der er karakteristiske for lægemiddelopdagelsesprogrammer, kræver avancerede beregningsmuligheder. HTBAC giver en sådan støtte gennem et fleksibelt mellemware -softwarelag, der muliggør adaptiv udførelse af algoritmer. I øjeblikket, HTBAC understøtter to algoritmer:forbedret sampling af molekylær dynamik med tilnærmelse af kontinuumopløsningsmiddel (ESMACS) og termodynamisk integration med forbedret sampling (TIES). ESMACS, en beregningsmæssigt billigere, men mindre streng metode end TIES, beregner bindestyrken af et lægemiddel til dets målprotein på basis af molekylære dynamiksimuleringer. Derimod, TIES sammenligner de relative bindingsaffiniteter for to forskellige lægemidler med det samme protein.
"ESMACS giver en hurtig kvantitativ tilgang, der er følsom nok til at bestemme bindingsaffiniteter, så vi kan fjerne dårlige bindemidler, mens TIES giver en mere præcis metode til at undersøge gode bindemidler, efterhånden som de forfines og forbedres, "sagde Jumana Dakka, et andet års ph.d. elev på Rutgers og medlem af gruppen RADICAL.
For at bestemme hvilken algoritme der skal udføres, HTBAC analyserer bindingsaffinitetsberegningerne ved runtime. Denne analyse informerer beslutninger om antallet af samtidige simuleringer, der skal udføres, og om stimuleringstrin skal tilføjes eller fjernes for hver undersøgt lægemiddelkandidat.
Sætter rammen på prøve
Jha's team demonstrated how HTBAC could provide insight from drug candidate data on a short timescale by reproducing results from a collaborative study between University College London and the London-based pharmaceutical company GlaxoSmithKline to discover drug compounds that bind to the BRD4 protein. Known to play a key role in driving cancer and inflammatory diseases, the BRD4 protein is a major target of bromodomain-containing (BRD) inhibitors, a class of pharmaceutical drugs currently being evaluated in clinical trials. The researchers involved in this collaborative study are focusing on identifying promising new drugs to treat breast cancer while developing an understanding of why certain drugs fail in the presence of breast cancer gene mutations.
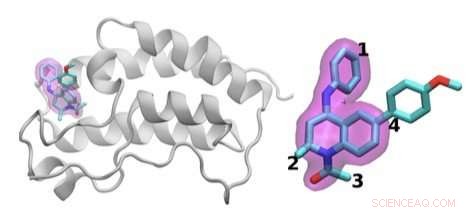
The scientists investigated the chemical structures of 16 drugs based on the same tetrahydroquinoline (THQ) scaffold. On the left is a cartoon of the BRD4 protein bound to one of these drugs; on the right is a molecular representation of a drug with the THQ scaffold highlighted in magenta. Regions that are chemically modified between the drugs investigated in this study are labeled 1 to 4. Typically, only a small change is made to the chemical structure of one drug to the next. This conservative approach makes it easier for researchers to understand why one drug is effective, whereas another is not. Kredit:Brookhaven National Laboratory
Jha and his team concurrently screened a group of 16 closely related drug candidates from the study by running thousands of computational sequences on more than 32, 000 computing cores. They ran the computations on the Blue Waters supercomputer at the National Center for Computing Applications, University of Illinois at Urbana-Champaign.
In a real drug design scenario, many more compounds with a wider range of chemical properties would need to be investigated. The team members previously demonstrated that the workload management layer and runtime system underlying HTBAC could scale to handle 10, 000 concurrent tasks.
"HTBAC could support the concurrent screening of different compounds at unprecedented scales—both in the number of compounds and computational resources used, " said Jha. "We showed that HTBAC has the ability to solve a large number of drug candidates in essentially the same amount of time it would take to solve a smaller set, assuming the number of processors increases proportionally with the number of candidates."
This ability is made possible through HTBAC's adaptive functionality, which allows it to execute the optimal algorithm depending on the properties of the drugs being investigated, improving the accuracy of the results and minimizing compute time.
"The lead optimization stage usually considers on the order of 10, 000 small molecules, " said Jha. "While experiment automation reduces the amount of time needed to calculate the binding affinities, HTBAC has the potential to cut this time (and cost) by an order of magnitude or more."
With HTBAC, TIES requires approximately 25, 000 central processing unit (CPU) core hours for a single prediction. At least a 250 million core hours would be needed for a large-scale study to support a pharmaceutical drug screening campaign, with a typical turnaround time of about two weeks. HTBAC could facilitate running studies requiring sustained usage of millions of core hours per day.
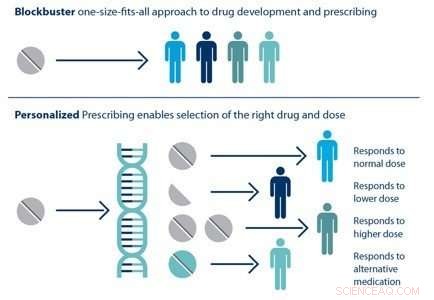
Individual patients respond differently to drugs. The ability to predict which treatment is best for a particular patient based on his or her genetic sequence is the goal of personalized medicine. Kredit:Brookhaven National Laboratory
When the University of College London–GlaxoSmithKline study concludes, Jha and his team hope to be given the experimental data on the tens of thousands of drug candidates, without knowing which candidate ended up being the best one. Med disse oplysninger, they could perform a blind test to determine whether HTBAC provides an improvement in compute time (for a given accuracy) over the existing automated methods for drug discovery. If necessary, they could then refine their methodology.
Applying scalable computing to precision medicine
HTBAC not only has the potential to improve the speed and accuracy of drug discovery in the pharmaceutical industry but also to improve individual patient outcomes in clinical settings. Using target proteins based on a patient's genetic sequence, HTBAC could predict a patient's response to different drug treatments. This personalized assessment could replace the traditional one-size-fits-all approach to medicine. For eksempel, such predictions could help determine which cancer patients would actually benefit from chemotherapy, avoiding unnecessary toxicity.
According to Jha, the computation time would have to be significantly reduced in order for physicians to clinically use HTBAC to treat their patients:"Our grand vision is to apply scalable computing techniques to personalized medicine. If we can use these techniques to optimize drugs and drug cocktails for each individual's unique genetic makeup on the order of a few days, we will be empowered to treat diseases much more effectively."
"Extreme-scale computing for precision medicine is an emerging area that CSI and Brookhaven at large have begun to tackle, " said CSI Director Kerstin Kleese van Dam. "This work is a great example of how technologies we originally developed to tackle DOE challenges can be applied to other domains of high national impact. We look forward to forming more strategic partnerships with other universities, pharmaceutical companies, and medical institutions in this important area that will transform the future of health care."
Sidste artikelEt år efter databrud:Atlanta-baserede Equifax løsnet
Næste artikelOpstartskubende softwaresystem til ændring af sundhedsadfærd
 Varme artikler
Varme artikler
-
 Afstemning finder, at kvinder er mere bekymrede for at miste job til handel og offshoring end kunsti…Kredit:CC0 Public Domain Så mange som 160 millioner kvinder rundt om i verden kan miste deres job i løbet af det næste årti på grund af virkningen af automatisering, og en nylig undersøgelse fra
Afstemning finder, at kvinder er mere bekymrede for at miste job til handel og offshoring end kunsti…Kredit:CC0 Public Domain Så mange som 160 millioner kvinder rundt om i verden kan miste deres job i løbet af det næste årti på grund af virkningen af automatisering, og en nylig undersøgelse fra -
 Ægteskab kun et klik væk for Chinas desperate enlige mændWebsites forbinder lovelorn kinesiske mænd med vietnamesiske kvinder, presset af fattigdom derhjemme til at gifte sig tusinder af miles væk Ægteskab med en vietnamesisk brud er bare et klik - og e
Ægteskab kun et klik væk for Chinas desperate enlige mændWebsites forbinder lovelorn kinesiske mænd med vietnamesiske kvinder, presset af fattigdom derhjemme til at gifte sig tusinder af miles væk Ægteskab med en vietnamesisk brud er bare et klik - og e -
 Næsten modellering af den menneskelige hjerne i en computerNeuroner i musens hjernebark rekonstrueret fra elektronmikroskopibilleder (grå). Hver nervecelle danner kontakter til tusindvis af andre celler. Forskere analyserer funktionerne i disse netværk ved hj
Næsten modellering af den menneskelige hjerne i en computerNeuroner i musens hjernebark rekonstrueret fra elektronmikroskopibilleder (grå). Hver nervecelle danner kontakter til tusindvis af andre celler. Forskere analyserer funktionerne i disse netværk ved hj -
 Dette nye atomur er så præcist, det kan bruges til at opdage mørkt stofNIST -fysiker Andrew Ludlow og kolleger opnåede nye rekorder for atomurpræstationer i en sammenligning af to ytterbium optiske gitterure. Lasersystemer, der bruges i begge ure, er synlige i forgrunden
Dette nye atomur er så præcist, det kan bruges til at opdage mørkt stofNIST -fysiker Andrew Ludlow og kolleger opnåede nye rekorder for atomurpræstationer i en sammenligning af to ytterbium optiske gitterure. Lasersystemer, der bruges i begge ure, er synlige i forgrunden
- Forskere identificerer en omkostningseffektiv metode til at fjerne forurenende stoffer fra kulstofna…
- Forskel mellem Plant & Animal Cell Division
- Reaktion af en kvantevæske til fotoekscitation af opløste partikler observeret for første gang
- Amazon til stille Alexas cackling
- Askehuggere kappes med tiden, før biller får dem alle
- Undersøgelse finder Jordens magnetfelt enklere, end vi troede