


Maskinindlæringsmetode forudsiger nøjagtigt metalliske defekter
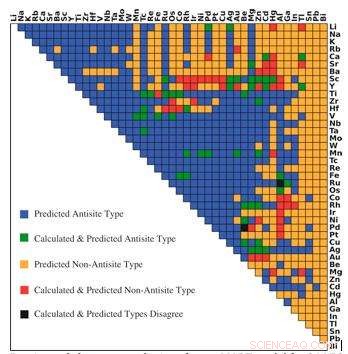
Dominerende defekttypeforudsigelser fra r-MART-model til 946 B2-type intermetallics. Farver angiver forholdet mellem forudsigelse og beregninger som vist i forklaringen. Kredit:Bharat Medasani, Berkeley Lab / PNNL
For første gang, forskere ved Lawrence Berkeley National Laboratory (Berkeley Lab) har bygget og uddannet maskinlæringsalgoritmer til at forudsige defektadfærd i visse intermetalliske forbindelser med høj nøjagtighed. Denne metode vil fremskynde forskning af nye avancerede legeringer og lette nye materialer til applikationer, der spænder over bilindustrien til rumfart og meget mere.
Deres resultater blev offentliggjort i december 2016 -udgaven af Natur beregningsmaterialer .
Materialer er aldrig kemisk rene og strukturelt fejlfrie. De indeholder næsten altid fejl, som spiller en vigtig rolle ved diktering af egenskaber. Disse mangler kan forekomme som ledige stillinger, som i det væsentlige er 'huller' i stoffets krystalstruktur, eller antisite defekter, som i det væsentlige er atomer placeret på det forkerte krystalsted. Forståelse af sådanne punktdefekter er afgørende for forskere, der designer materialer, fordi de kan have en dramatisk effekt på lang tid strukturel stabilitet og styrke.
Traditionelt set forskere har brugt en beregningsmæssig kvantemekanisk metode kendt som densitetsfunktionelle beregninger til at forudsige, hvilke slags defekter der kan dannes i en given struktur, og hvordan de påvirker materialets egenskaber. Selvom det er effektivt, denne fremgangsmåde er meget beregningsmæssigt dyr at udføre for punktdefekter, der begrænser omfanget af sådanne undersøgelser.
"Densitetsfunktionelle beregninger fungerer godt, hvis du modellerer en lille enhed, men hvis du vil gøre din modelcelle større, stiger beregningsevnen, der kræves for at gøre dette, betydeligt, "siger Bharat Medasani, en tidligere postdoc i Berkeley Lab og hovedforfatter af NPJ -papiret. "Og fordi det er beregningsmæssigt dyrt at modellere fejl i et enkelt materiale, Det er ikke muligt at foretage denne form for brutal kraftmodellering for titusinder af materialer. "
For at overvinde disse computerudfordringer, Medasani og hans kolleger udviklede og uddannede maskinlæringsalgoritmer til at forudsige punktdefekter i intermetalliske forbindelser, med fokus på den meget observerede B2 -krystalstruktur. I første omgang, de valgte en prøve på 100 af disse forbindelser fra Materials Project Database og kørte tæthedsfunktionelle beregninger på supercomputere ved National Energy Research Scientific Computing Center (NERSC), en DOE Office of Science User Facility på Berkeley Lab, at identificere deres fejl.
Fordi de havde en lille dataprøve at arbejde ud fra, Medasani og hans team brugte en skovtilgang kaldet gradient boosting til at udvikle deres machine learning -metode til en høj nøjagtighed. I denne tilgang blev yderligere maskinlæringsmodeller bygget successivt og kombineret med tidligere modeller for at minimere forskellen mellem modellernes forudsigelser og resultaterne fra tæthedsfunktionelle beregninger. Forskerne gentog processen, indtil de opnåede en høj nøjagtighed i deres forudsigelser.
"Dette værk er i det væsentlige et bevis på konceptet. Det viser, at vi kan køre tæthedsfunktionelle beregninger for et par hundrede materialer, derefter træne maskinlæringsalgoritmer til præcist at forudsige punktdefekter for en meget større gruppe materialer, "siger Medasani, som nu er postdoktor ved Pacific Northwest National Laboratory.
"Fordelen ved dette arbejde er nu, at vi har en beregningsmæssigt billig maskinlæringsmetode, der hurtigt og præcist kan forudsige punktdefekter i nye intermetalliske materialer" siger Andrew Canning, en Berkeley Lab Computational Scientist og medforfatter på npj-papiret. "Vi behøver ikke længere at køre meget dyre første principberegninger for at identificere defekte egenskaber for hver ny metallisk forbindelse."
"Dette værktøj gør det muligt for os at forudsige metalliske defekter hurtigere og mere robust, hvilket igen vil fremskynde materialedesign, siger Kristin Persson, en Berkeley Lab -forsker og direktør for materialeprojektet, et initiativ, der har til formål drastisk at reducere den tid, der er nødvendig for at opfinde nye materialer ved at tilbyde åben webbaseret adgang til beregnede oplysninger om kendte og forudsagte materialer. Som en forlængelse af dette arbejde er der udviklet en open source Python -værktøjskasse til modelleringspunktfejl i halvledere og isolatorer (PyCDT).
Sidste artikelNy teori til at forklare, hvorfor soloverfladen roterer langsommere end dens kerne
Næste artikelLæringens termodynamik
 Varme artikler
Varme artikler
-
 Atomic jet-den første linse til ekstremt ultraviolet lys udvikletFokusering af en XUV -stråle af en atomstråle, der bruges som en linse. Kredit:MBI Berlin Forskere fra Max Born Institute (MBI) har udviklet det første brydningsobjektiv, der fokuserer ekstreme ul
Atomic jet-den første linse til ekstremt ultraviolet lys udvikletFokusering af en XUV -stråle af en atomstråle, der bruges som en linse. Kredit:MBI Berlin Forskere fra Max Born Institute (MBI) har udviklet det første brydningsobjektiv, der fokuserer ekstreme ul -
 Søger efter axion mørkt stof med en ny detektionsenhedKredit:Yale University En detektionsenhed designet og bygget på Yale indsnævrer søgningen efter mørkt stof i form af aksioner, en teoretiseret subatomær partikel, der kan udgøre så meget som 80% a
Søger efter axion mørkt stof med en ny detektionsenhedKredit:Yale University En detektionsenhed designet og bygget på Yale indsnævrer søgningen efter mørkt stof i form af aksioner, en teoretiseret subatomær partikel, der kan udgøre så meget som 80% a -
 En elektrisk kontakt til magnetisme(Top) Skematisk felteffekttransistor baseret på ultratynd ferromagnetisk halvleder Cr2Ge2Te6. Materialet er dækket med en iongel for at forstærke felteffekten. (Bund) Magneto-modstand (MR) med stigend
En elektrisk kontakt til magnetisme(Top) Skematisk felteffekttransistor baseret på ultratynd ferromagnetisk halvleder Cr2Ge2Te6. Materialet er dækket med en iongel for at forstærke felteffekten. (Bund) Magneto-modstand (MR) med stigend -
 Sådan konverteres tilspidsning til graderTaper er et gradvis fald i højde eller bredde. Det udtrykkes normalt i inches over 1 fod. En ingeniør kræver muligvis en tilspidsning på 2,5 inches per fod, hvilket betyder et fald på 2,5 inches for h
Sådan konverteres tilspidsning til graderTaper er et gradvis fald i højde eller bredde. Det udtrykkes normalt i inches over 1 fod. En ingeniør kræver muligvis en tilspidsning på 2,5 inches per fod, hvilket betyder et fald på 2,5 inches for h
- Ombygning af ruiner for at bevare landdistrikternes arv
- Sådan bruges en protractor til at måle en trekant
- Kobes smart city-projekt begynder under jorden
- Såning af hvede tidligere kan hjælpe med at øge udbyttet i Indien
- Gode steder at finde tomme Aluminium Cans
- Hvad er din idé om at 3D-udskrive på månen-for at få det til at føle sig som hjemme?