


Steady-state densitet funktionel teori
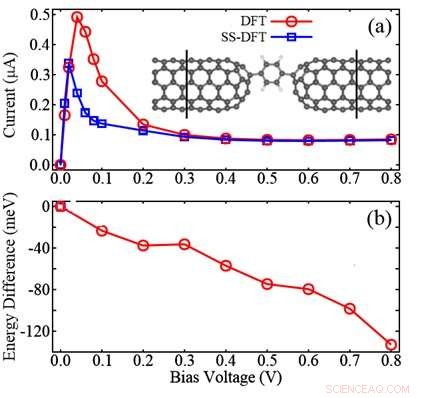
Figur viser sammenligningen mellem SS-DFT og meget udbredte DFT-metoder for en molekylær anordning bestående af to carbon nanorør (CNT) elektroder og et benzenmolekyle imellem:(a) beregnede IV-kurver; (b) energiforskellen beregnet ved at trække DFT-energien fra SS-DFT-en. Figuren viser, at SS-DFT forudsiger den energisk mere stabile transporttilstand med lavere elektriske strømme sammenlignet med den DFT-baserede metode. Kredit:Zhang Chun
NUS -beregningsforskere har udviklet en ny version af densitetsfunktionelle teori (DFT) til undersøgelse af nanoskalaenheder.
Elektroniske enheder bliver mindre og indeholder større funktionalitet. Dette er muliggjort ved at reducere størrelsen på de elektroniske komponenter. Når deres størrelse falder, egenskaberne ved disse molekylære enheder bliver meget mere følsomme over for deres ydre miljø. Beregningsmetoder er nødvendige for at simulere og forudsige egenskaberne af sådanne små enheder. En af dem er densitetsfunktionelle teori. Disse metoder er udviklet ud fra de første principper, omfattende grundlæggende og grundlæggende viden, som vi allerede kender. NUS-beregningsforskere har forfinet teorien for at tage højde for ikke-ligevægtseffekter, der er til stede under driften af enhederne (f.eks. Når et batteri er forbundet til et kvantesystem). Dette fører til en mere præcis og præcis forudsigelse.
Prof ZHANG Chun og hans ph.d. studerende, LIU Shuanglong sammen med forsker, Dr Argo NURBAWONO, fra Institut for Fysik, NUS har udviklet en mere generel version af den populære og meget udbredte densitetsfunktionelle teori (DFT), der kan anvendes til steady-state ikke-ligevægtssituationer. De indførte en ekstra grad af frihed, kendt som ikke-ligevægts elektrontæthed, ind i modellerne med de første principper. Dette tager højde for de bias-inducerede ikke-ligevægtseffekter, når en molekylær enhed fungerer under en begrænset bias. Denne nye version af teorien er kendt som steady-state DFT (SS-DFT).
Forskerne har vist, at den meget udbredte DFT i princippet ikke er korrekt i et steady-state ikke-ligevægtsscenario. I en sådan situation, to forskellige parametre, den samlede elektrontæthed og tætheden af strømførende elektroner er nødvendige for at bestemme egenskaberne for det tilsvarende ikke-ligevægtssystem. Den nye teori er blevet implementeret i det spanske initiativ til elektroniske simuleringer med tusindvis af atomer (SIESTA) beregningspakke til undersøgelse af elektroniske/ transportegenskaber ved forskellige enheder i molekylær skala.
SS-DFT giver et pålideligt teoretisk værktøj til forståelse og fremtidigt design af nye molekylære enheder med forbedret funktionalitet. Den SS-DFT-baserede beregningspakke bruges nu af mange forskningsgrupper over hele verden. Det bruges til at forklare spændende transportfænomener observeret eksperimentelt på molekylært niveau og til at designe nye typer molekylære enheder.
Forskerne planlægger at udvide teoriens anvendelighed ved at inkludere andre fysiske effekter, såsom elektron-fonon-interaktioner og spin-orbitalkobling. De agter også at forbedre beregningseffektiviteten, så den kan bruges til at modellere store systemer omkring 1, 000 atomer.
 Varme artikler
Varme artikler
-
 Forskning forbinder elastodynamiske og elektromagnetiske bølgefænomenerDette skema viser det heterogene materiale, der samtidig selektivt blokerer lydbølger, men transmitterer lys, eller mere generelt, elektromagnetiske bølger. Kredit:Jaeuk Kim Forestil dig fremskrid
Forskning forbinder elastodynamiske og elektromagnetiske bølgefænomenerDette skema viser det heterogene materiale, der samtidig selektivt blokerer lydbølger, men transmitterer lys, eller mere generelt, elektromagnetiske bølger. Kredit:Jaeuk Kim Forestil dig fremskrid -
 Laserdrevet spin-dynamik i ferrimagneter:Hvordan flyder vinkelmomentet?I starten, Gd besidder ingen vinkelmoment (L =0), og der ses ingen akkumulering under demagnetiseringen, efter at laserpulsen har ramt prøven på tidspunktet nul. I Fe, både S og L falder i samme hasti
Laserdrevet spin-dynamik i ferrimagneter:Hvordan flyder vinkelmomentet?I starten, Gd besidder ingen vinkelmoment (L =0), og der ses ingen akkumulering under demagnetiseringen, efter at laserpulsen har ramt prøven på tidspunktet nul. I Fe, både S og L falder i samme hasti -
 Anvendelse af kvante-urenhedsteori til kvantefluider af lysSpektroskopisk signatur af topunkter, mangekroppskorreleret tilstand. Til venstre:I mangel af pumpning. Til højre:Med pumpning. Kredit:FLEET En Monash-ledet undersøgelse udvikler en ny tilgang til
Anvendelse af kvante-urenhedsteori til kvantefluider af lysSpektroskopisk signatur af topunkter, mangekroppskorreleret tilstand. Til venstre:I mangel af pumpning. Til højre:Med pumpning. Kredit:FLEET En Monash-ledet undersøgelse udvikler en ny tilgang til -
 Forskere udvikler akustisk metamateriale, der annullerer lydDet matematisk udformede, 3D-printet akustisk metamateriale er formet på en sådan måde, at det sender indgående lyde tilbage til, hvor de kom fra, Siger Ghaffarivardavagh og Zhang. Inde i den ydre rin
Forskere udvikler akustisk metamateriale, der annullerer lydDet matematisk udformede, 3D-printet akustisk metamateriale er formet på en sådan måde, at det sender indgående lyde tilbage til, hvor de kom fra, Siger Ghaffarivardavagh og Zhang. Inde i den ydre rin
- Rumvandrende astronauter forbereder sig på ankomsten af russisk laboratorium i 2021
- Hvad er de strukturelle dele af de lange ben i kroppen?
- Bakterier producerer guld ved at fordøje giftige metaller
- Modellering af magma for at finde kobber
- Gamle bønder skånede os fra gletsjere, men ændrede dybtgående jordens klima
- Forskere afslører in situ manipulation af aktivt guld-titandioxid-interface