


Pepsi-SAXS:Ny metode til proteinanalyse, der er 50 gange hurtigere end analoger

Pepsi-SAXS:Ny metode til proteinanalyse, der er 50 gange hurtigere end analoger. Kredit:MIPT
Pepsi-SAXS er en ny, højeffektiv metode til beregning af røntgenspredningsprofiler, som er nødvendige for analyse af proteinmolekyler i opløsningstilstand. Metoden blev skabt af forskere fra Université Grenoble Alpes og MIPT, ledet af Sergei Grudinin. Holdet testede deres metode, og resultaterne blev offentliggjort af International Union of Crystallography i dets tidsskrift Acta Crystallographica Afsnit D:Strukturel biologi .
Proteiner har kompleks struktur og ekstremt lille størrelse, i størrelsesordenen flere nanometer. For at studere dem, forskere skal finde på usædvanlige metoder, fordi proteinprøver alt for let ødelægges og deres egenskaber ændres i eksperimenter. Viden om biomolekylers strukturer og funktionelle mekanismer gør det muligt at udvikle nye lægemidler ikke ved forsøg og fejl - teknisk kaldet high throughput screening - men på en mere fokuseret måde.
En af de teknikker, der bruges til at studere proteiner, er analysen af røntgenstråler spredt fra dem. Forskere skal bruge røntgenstråler og ikke almindeligt lys for at zoome ind på individuelle atomer med en karakteristisk størrelse i størrelsesordenen 0,1 nanometer. Jo mindre objektet er, jo kortere lysets bølgelængde skal bruges til at observere det. Synligt lys omfatter bølgelængder mellem 400 og 700 nanometer. røntgenstråler, på den anden side, har en meget kortere bølgelængde og kan dermed bruges til at undersøge molekylære strukturer.
"Den nye metode giver os mulighed for at plotte spredningskurver effektivt og præcist, og analysere den tredimensionelle struktur af en prøve, " siger MIPT-studerende Maria Garkavenko, medforfatter til papiret. "Blandt andet, Pepsi-SAXS øger modelleringseffektiviteten og nøjagtigheden af tredimensionel makromolekylestrukturforudsigelse."
Lille-vinklet røntgenspredning, eller SAXS, er en eksperimentel teknik, der involverer spredning af røntgenstråler fra en prøve og derefter opsamling af dem i meget små vinkler. Som resultat, et plot af spredt røntgenstråleintensitet som funktion af indfaldsvinklen opnås. Ved at bruge dette plot, en proteinprøve kan sammenlignes med andre prøver i den eksperimentelle database for at bestemme dens struktur og egenskaber.
Sammenlignet med andre teknikker, der bruges til at bestemme prøvestruktur, SAXS er meget enklere og billigere. Det kræver kun et minimum af prøveforberedelse, og proteinerne behøver ikke at blive frosset eller krystalliseret. Prøverne studeres i opløsning og i deres funktionelle tilstand. Dette gør resultaterne meget mere pålidelige, fordi prøveforberedelse nogle gange kan ændre et proteins tilstand og egenskaber. En anden vigtig fordel ved metoden er, at den er ikke-destruktiv, hvilket betyder, at den eksperimentelle prøve forbliver stort set upåvirket af røntgenstråler.
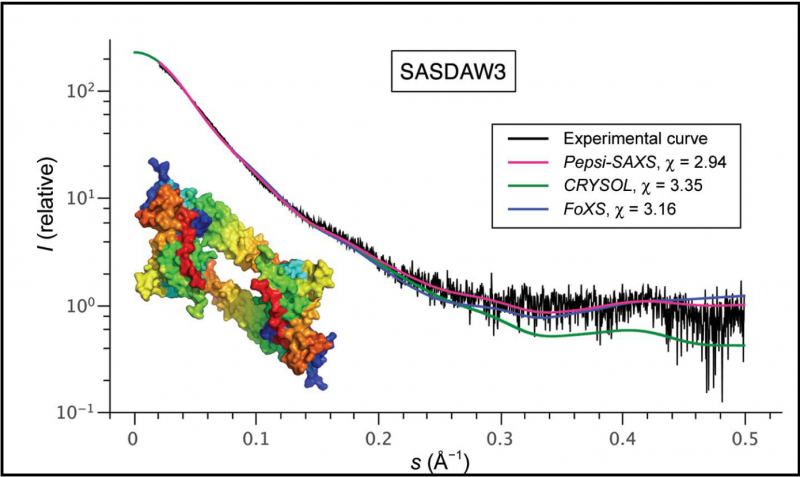
Figur 1 viser resultaterne af en række eksperimenter, som sammenlignede Pepsi-SAXS med to af de aktuelt anvendte beregningsmetoder ved at anvende dem på den samme prøve (SASDAW3) fra SASBDB-databasen. Gennemsnitlig spredt intensitet er plottet som en funktion af spredningsvinklen. Fejlen χ² i beregningsmodellen er den laveste i tilfældet med Pepsi-SAXS, som er resultatet af en mere præcis repræsentation af hydreringsskallen. Kredit:S. Grudinin, M. Garkavenko og A. Kazennov
Men indtil for nylig, SAXS havde en stor ulempe:Metoden var beregningsintensiv, hvilket betød, at det ikke kunne bruges, hvis antallet af eksperimenter var betydeligt. Det tog timer at behandle resultaterne af kun ét eksperiment. I første omgang, antallet af beregninger var direkte proportionalt med kvadratet af antallet af atomer i prøven, sidstnævnte tal overstiger sædvanligvis tusind. Imidlertid, i 1970'erne, Heinrich Stuhrmann, en tysk forsker, kom med en idé, der forenklede beregningerne. Han foreslog, at spredning på molekylære forbindelser beskrives med funktioner af en bestemt art kaldet sfæriske harmoniske. Denne tilgang viste sig at være en succes. I årenes løb, en række beregningsværktøjer til analyse af SAXS-data blev skabt. Vigtige bidrag til deres udvikling blev givet af forskere med en sovjetisk videnskabelig baggrund, herunder Dmitri Svergun (arbejder i øjeblikket i Hamborg), som skrev ATSAS-softwarepakken til SAXS-dataanalyse i biologisk makromolekyleforskning. Forskerne i undersøgelsen, der er rapporteret her, undersøgte flere beregningsmetoder og sammenlignede dem med deres egen teknik.
"Pepsi-SAXS står for 'polynomielle udvidelser af proteinstrukturer og interaktioner' og 'lillevinklet røntgenspredning'. Det er en adaptiv metode til hurtig og nøjagtig beregning af småvinklede røntgenstrålespredningsprofiler, " forklarer MIPT Ph.D.-studerende Andrei Kazennov, medforfatter til papiret. "Pepsi-SAXS kan tilpasses til størrelsen af en given prøve og opløsningen af eksperimentelle data."
Forskerne skabte også en effektiv model af hydreringsskallen - et lag af vandmolekyler, der omgiver proteiner i opløsning - og inkorporerede det i deres software, øger metodens nøjagtighed.
"Vores metode er blevet valideret på et stort datasæt fra BioIsis og SASBDB, de to største biologiske databaser, " siger Sergei Grudinin, der overvågede forskningen. "Vi har vist, at Pepsi-SAXS er fem til 50 gange hurtigere end de tidligere anvendte metoder, nemlig CRYSOL, FoXS, og den tredimensionelle Zernike-teknik implementeret i SAStbx-pakken. På samme tid, nøjagtigheden er på niveau med dem."
Forskerne lagde særlig vægt på analysen af de opnåede resultater, som blev sammenlignet med de eksperimentelle data.
Proteinforskning har grundlæggende betydning for vores forståelse af de grundlæggende processer, der ligger til grund for livet, såvel som til udvikling af lægemidler, behandlinger, og organiske materialer, herunder kunstige organer. Det nye værktøj præsenteret af forfatterne kan betyde 50 gange hurtigere fremskridt på disse områder.
 Varme artikler
Varme artikler
-
 Ny tilgang kunne gøre HVAC varmevekslere fem gange mere effektiveKredit:Brown University Forskere fra Tsinghua University og Brown University har opdaget en enkel måde at give et stort løft til turbulent varmeudveksling, en metode til varmetransport, der i vid
Ny tilgang kunne gøre HVAC varmevekslere fem gange mere effektiveKredit:Brown University Forskere fra Tsinghua University og Brown University har opdaget en enkel måde at give et stort løft til turbulent varmeudveksling, en metode til varmetransport, der i vid -
 Forskere når milepæl i 3D-laserskrivning i bulk siliciumEksperimentel opsætning af brug af 60-femtosekund laserpulser til laserskrivning i silicium. Kredit:Chanal et al. Udgivet i Naturkommunikation (Phys.org) - Det har taget mere end 20 år, men fors
Forskere når milepæl i 3D-laserskrivning i bulk siliciumEksperimentel opsætning af brug af 60-femtosekund laserpulser til laserskrivning i silicium. Kredit:Chanal et al. Udgivet i Naturkommunikation (Phys.org) - Det har taget mere end 20 år, men fors -
 Forskere knækker en varig fysikgådeTobias Schneider og Florian Reetz. Kredit:Ecole Polytechnique Federale de Lausanne (EPFL) I årtier, fysikere, ingeniører og matematikere har undladt at forklare et bemærkelsesværdigt fænomen inden
Forskere knækker en varig fysikgådeTobias Schneider og Florian Reetz. Kredit:Ecole Polytechnique Federale de Lausanne (EPFL) I årtier, fysikere, ingeniører og matematikere har undladt at forklare et bemærkelsesværdigt fænomen inden -
 Opnå UV -ikke -linearitet med en bred båndgab halvlederbølgelederSkematisk af AlInGaN polariton bølgelederstruktur. Kredit:Dr. Paul Walker, University of Sheffield. Området ultrahurtig ikke -lineær fotonik er nu blevet omdrejningspunktet for talrige undersøgels
Opnå UV -ikke -linearitet med en bred båndgab halvlederbølgelederSkematisk af AlInGaN polariton bølgelederstruktur. Kredit:Dr. Paul Walker, University of Sheffield. Området ultrahurtig ikke -lineær fotonik er nu blevet omdrejningspunktet for talrige undersøgels
- Forskellen mellem mekanisk og kinetisk energi
- Den fantastiske mangfoldighed – og mulige tilbagegang – af svampe og andre svampe
- Kunne gårsdagens Jord indeholde spor til at lave morgendagens medicin?
- Spin-to-charge-konvertering opnår 95 % samlet qubit-udlæsningsnøjagtighed
- Et spørgsmål om tillid:skal chefer være i stand til at spionere på arbejdere, selv når de arbej…
- Toyota vil bygge testbane til selvkørende biler