


Sådan måles et molekyls energi ved hjælp af en kvantecomputer
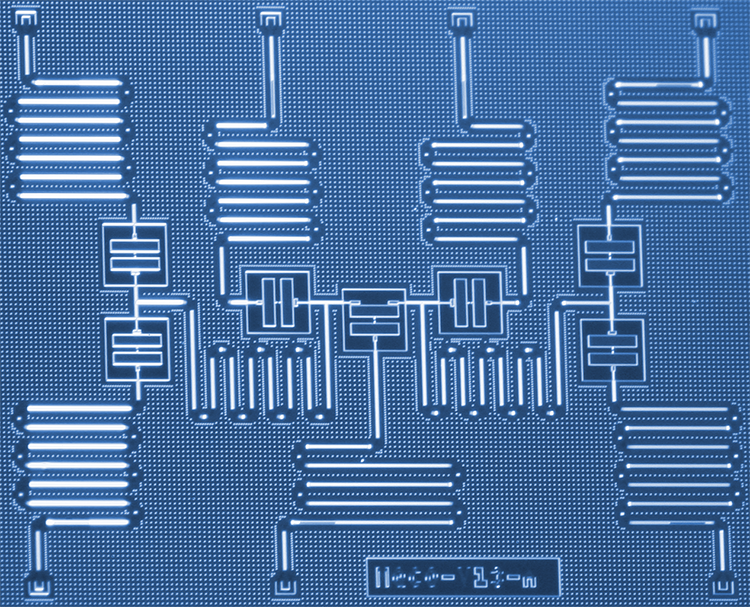
IBM -forskere har udviklet en ny tilgang til at simulere molekyler på en kvantecomputer, der en dag kan hjælpe med at revolutionere kemi og materialevidenskab. Forskerne har med succes brugt seks qubits på en specialbygget syv-qubit kvanteprocessor til at løse problemerne med molekylstruktur for berylliumhydrid (BeH2)-det største molekyle, der er simuleret på en kvantecomputer til dato. Resultaterne viser en udforskningsvej for kortsigtede kvantesystemer for at forbedre vores forståelse af komplekse kemiske reaktioner, der kan føre til praktiske anvendelser. Kredit:Kandala et al .; Natur
Simulering af molekyler på kvantecomputere blev bare meget lettere med IBMs superledende kvantehardware. I en nylig forskningsartikel offentliggjort i Natur , Hardwareeffektiv variation af Quantum Eigensolver til små molekyler og kvantemagneter, vi implementerer en ny kvantealgoritme, der effektivt kan beregne den laveste energistatus for små molekyler. Ved at kortlægge den elektroniske struktur af molekylære orbitaler på en delmængde af vores specialbyggede syv qubit kvanteprocessor, vi studerede molekyler, der tidligere ikke var udforsket med kvantecomputere, herunder lithiumhydrid (LiH) og berylliumhydrid (BeH2). Den særlige kodning fra orbitaler til qubits studeret i dette arbejde kan bruges til at forenkle simuleringer af endnu større molekyler, og vi forventer muligheden for at udforske sådanne større simuleringer i fremtiden, når kvanteberegningseffekten (eller "kvantevolumen") i IBM Q -systemer er steget.
Mens BeH2 til dato er det største molekyle, der nogensinde er simuleret af en kvantecomputer, den betragtede model af selve molekylet er stadig enkel nok til, at klassiske computere kan simulere nøjagtigt. Dette gjorde det til en testcase at skubbe grænserne for, hvad vores syv qubit -processor kunne opnå, yderligere vores forståelse af kravene for at forbedre nøjagtigheden af vores kvantesimuleringer, og lægge de grundlæggende elementer, der er nødvendige for at udforske sådanne molekylære energistudier.
De bedste simuleringer af molekyler i dag køres på klassiske computere, der bruger komplekse omtrentlige metoder til at estimere den laveste energi af en molekylær Hamiltonian. En "Hamiltonian" er en kvantemekanisk energioperatør, der beskriver interaktionerne mellem alle elektronorbitaler og kerner i de konstituerende atomer. Den "laveste energi" -tilstand for den molekylære Hamiltonian dikterer molekylets struktur og hvordan den vil interagere med andre molekyler. Sådanne oplysninger er afgørende for kemikere til at designe nye molekyler, reaktioner, og kemiske processer til industrielle applikationer.
Qubit:Orbital
Selvom vores syv qubit kvanteprocessor ikke er fuldstændigt fejlkorrigeret og fejltolerant, sammenhængstiderne for de enkelte qubits varer cirka 50 µs. Det er derfor virkelig vigtigt at bruge en meget effektiv kvantealgoritme til at få mest muligt ud af vores dyrebare kvantesammenhæng og forsøge at forstå molekylære strukturer. Algoritmen skal være effektiv med hensyn til antallet af anvendte qubits og antallet af udførte kvanteoperationer.

Anvendelse til kvantekemi. a – c, Eksperimentelle resultater (sorte fyldte cirkler), nøjagtige energioverflader (stiplede linjer) og tæthedsplotter (skygge; se farveskalaer) af resultater fra numeriske simuleringer, for flere interatomiske afstande for H2 (a), LiH (b) og BeH2 (c). De præsenterede eksperimentelle og numeriske resultater er for kredsløb med dybde d =1. Fejlstængerne på de eksperimentelle data er mindre end markørernes størrelse. Tæthedsplottene opnås fra 100 numeriske resultater ved hver interatomisk afstand. De øverste indsatser i hvert panel fremhæver de qubits, der blev brugt til eksperimentet og krydsresonansporte (pile, mærket CRc – t; hvor 'c' betegner kontrolkvbit og 't' målqubit), der udgør UENT. De nederste indsatser er repræsentationer af den molekylære geometri (ikke i målestok). For alle de tre molekyler, afvigelsen af de eksperimentelle resultater fra de nøjagtige kurver forklares godt af de stokastiske simuleringer. Kredit:Kandala et al .; Natur
Vores skema står i kontrast til tidligere undersøgte kvantesimuleringsalgoritmer, som fokuserer på at tilpasse klassiske molekylære simuleringsordninger til kvantehardware - og dermed ikke effektivt tage hensyn til de begrænsede omkostninger ved nuværende realistiske kvanteenheder.
Så i stedet for at tvinge klassiske computermetoder til kvantehardware, vi har vendt tilgangen og spurgt:hvordan kan vi udtrække den maksimale kvanteberegningseffekt ud af vores syv qubit -processor?
Vores svar på dette kombinerer en række hardware-effektive teknikker til at angribe problemet:
- Først, et molekyls fermioniske Hamiltonian omdannes til en qubit Hamiltonian, med en ny effektiv kortlægning, der reducerer antallet af qubits, der kræves i simuleringen.
- Et hardwareffektivt kvantekredsløb, der udnytter de naturligt tilgængelige gateoperationer i kvanteprocessoren, bruges til at forberede teststater for Hamilton.
- Kvanteprocessoren køres til forsøgsstanden, og der udføres målinger, der giver os mulighed for at evaluere energien i den forberedte forsøgstilstand.
- De målte energiværdier føres til en klassisk optimeringsrutine, der genererer det næste kvantekredsløb til at drive kvanteprocessoren til, for yderligere at reducere energien.
- Iterationer udføres, indtil den laveste energi er opnået til den ønskede nøjagtighed.
Med fremtidige kvanteprocessorer, der vil have mere kvantevolumen, vi vil være i stand til at undersøge kraften i denne tilgang til kvantesimulering for stadig mere komplekse molekyler, der ligger ud over klassiske computermuligheder. Evnen til at simulere kemiske reaktioner præcist, er ledende i bestræbelserne på at opdage nye lægemidler, gødning, endda nye bæredygtige energikilder.
De eksperimenter, vi beskriver i vores papir, blev ikke kørt på vores i øjeblikket offentligt tilgængelige fem qubit og 16 qubit processorer på skyen. Men udviklere og brugere af IBM Q -oplevelsen kan nu få adgang til Jupyter -notebooks med kvantekemi på QISKit github -repoen. På systemet med fem qubit, brugere kan udforske jordtilstandenergisimulering for de små molekyler hydrogen og LiH. Bærbare computere til større molekyler er tilgængelige for dem med beta-adgang til den opgraderede 16-qubit processor.
 Varme artikler
Varme artikler
-
 Overraskende resultat chokerer forskere, der studerer spinNeutroner, der produceres, når en spin-justeret (polariseret) proton kolliderer med en anden proton, kommer ud med en let højre-skæv præference. Men når den polariserede proton kolliderer med en meget
Overraskende resultat chokerer forskere, der studerer spinNeutroner, der produceres, når en spin-justeret (polariseret) proton kolliderer med en anden proton, kommer ud med en let højre-skæv præference. Men når den polariserede proton kolliderer med en meget -
 Variationer i vibrationer i siliciumbjælker skaber en følsom måde at måle trykændringer påMohammad Younis (tilbage) og Nouha Alcheikh diskuterer den optimale tykkelse for strålen i deres tryksensor. Kredit:KAUST En mikrometer-skala, lavt strømforbrug tryksensor er blevet udviklet af KA
Variationer i vibrationer i siliciumbjælker skaber en følsom måde at måle trykændringer påMohammad Younis (tilbage) og Nouha Alcheikh diskuterer den optimale tykkelse for strålen i deres tryksensor. Kredit:KAUST En mikrometer-skala, lavt strømforbrug tryksensor er blevet udviklet af KA -
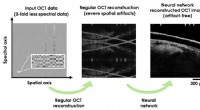 Deep learning forbedrer billedrekonstruktion i optisk kohærenstomografi ved brug af færre dataDeep learning forbedrer billedrekonstruktion i optisk kohærenstomografi ved brug af væsentligt færre spektrale data. Kredit:Ozcan Lab @UCLA. Optisk kohærenstomografi (OCT) er en ikke-invasiv bille
Deep learning forbedrer billedrekonstruktion i optisk kohærenstomografi ved brug af færre dataDeep learning forbedrer billedrekonstruktion i optisk kohærenstomografi ved brug af væsentligt færre spektrale data. Kredit:Ozcan Lab @UCLA. Optisk kohærenstomografi (OCT) er en ikke-invasiv bille -
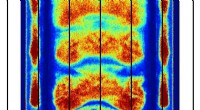 Hvorfor lyser pulsarer:Et halvt århundrede gammelt mysterium er løstDen simulerede densitetsfordeling af elektron-positronplasma nær overfladen af en neutronstjerne (vist med gråt i bunden af plottet). Rødere områder repræsenterer en højere densitet af elektron-po
Hvorfor lyser pulsarer:Et halvt århundrede gammelt mysterium er løstDen simulerede densitetsfordeling af elektron-positronplasma nær overfladen af en neutronstjerne (vist med gråt i bunden af plottet). Rødere områder repræsenterer en højere densitet af elektron-po
- Små partikler stiger i luften med ethanol-til-benzin switch
- Flere af den kinesiske befolkning vil blive udsat for hedebølger
- Sådan konverteres grader til tommer eller millimeter
- Tæt hus, immigration flytter pres på Seattles sorte kvarterer, sociolog finder
- Undersøgelse bruger terahertz laserpulser til at afsløre ultrahurtig kobling af atomskala mønstre
- Hvad er en Radian?