


Quantum forudsigelser
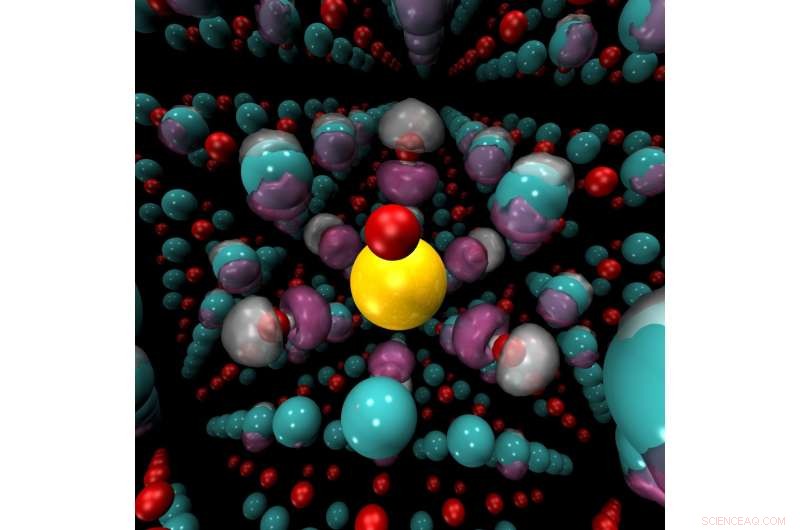
Mekanisk belastning, tryk- eller temperaturændringer eller tilføjelse af kemiske dopingmidler kan forårsage en pludselig skift fra isolator til leder i materialer som nikkeloxid (billedet her). Nikkelioner (blå) og iltioner (røde) omgiver en dopantion af kalium (gul). Quantum Monte Carlo -metoder kan præcist forudsige regioner, hvor ladningstæthed (lilla) vil akkumulere i disse materialer. Kredit:Anouar Benali, Argonne National Laboratory
At løse et komplekst problem hurtigt kræver omhyggelige afvejninger - og simulering af materialers adfærd er ingen undtagelse. For at få svar, der muligvis forudsiger molekylært arbejde, forskere skal bytte i matematiske tilnærmelser, der fremskynder beregningen for nøjagtighedens regning.
Men magnetisme, elektrisk ledningsevne og andre egenskaber kan være ret sarte, siger Paul R.C. Kent fra Department of Energy's (DOE's) Oak Ridge National Laboratory. Disse egenskaber afhænger af kvantemekanik, bevægelser og interaktioner mellem utallige elektroner og atomer, der danner materialer og bestemmer deres egenskaber. Forskere, der studerer sådanne træk, skal modellere store grupper af atomer og molekyler frem for blot nogle få. Dette problems kompleksitet kræver øget beregningsværktøjers effektivitet og nøjagtighed.
Det er her en metode kaldet quantum Monte Carlo (QMC) modellering kommer ind. Mange andre teknikker tilnærmer elektronernes adfærd som et samlet gennemsnit, for eksempel, frem for at betragte dem individuelt. QMC muliggør regnskab for den enkelte adfærd for alle elektronerne uden større tilnærmelser, reducere systematiske fejl i simuleringer og producere pålidelige resultater, siger Kent.
Kents interesse for QMC går tilbage til hans ph.d. forskning ved Cambridge University i 1990'erne. Hos ORNL, han vendte for nylig tilbage til metoden, fordi fremskridt inden for både supercomputerhardware og algoritmer havde gjort det muligt for forskere at forbedre dens nøjagtighed.
"Vi kan lave nye materialer og en bredere brøkdel af elementer på tværs af det periodiske system, "Siger Kent." Endnu vigtigere, vi kan begynde at lave nogle af materialerne og egenskaberne, hvor de mere omtrentlige metoder, som vi bruger dagligt, bare er upålidelige."
Selv med disse fremskridt, simuleringer af disse typer materialer, dem, der indeholder op til et par hundrede atomer og tusinder af elektroner, kræver beregningstunge løft. Kent leder et DOE Basic Energy Sciences Center, Center for Predictive Simulations of Functional Materials (CPSFM), der omfatter forskere fra ORNL, Argonne National Laboratory, Sandia National Laboratories, Lawrence Livermore National Laboratory, University of California, Berkeley og North Carolina State University.
Deres arbejde understøttes af en DOE Innovative and Novel Computational Impact on Theory and Experiments (INCITE) tildeling af 140 millioner processortimer, delt mellem Oak Ridge Leadership Computing Facility's Titan og Argonne Leadership Computing Facilitys Mira supercomputere. Begge computercentre er DOE Office of Science -brugerfaciliteter.
For at tage QMC til det næste niveau, Kent og kolleger starter med materialer som vanadiumdioxid, der viser usædvanlig elektronisk adfærd. Ved køligere temperaturer, dette materiale isolerer mod strømmen af elektricitet. Men ved lige over stuetemperatur, vanadiumdioxid ændrer pludselig dets struktur og adfærd.
Pludselig bliver dette materiale metallisk og leder elektricitet effektivt. Forskere forstår stadig ikke nøjagtigt, hvordan og hvorfor dette sker. Faktorer som mekanisk belastning, tryk eller doping af materialerne med andre elementer forårsager også denne hurtige overgang fra isolator til leder.
Imidlertid, hvis forskere og ingeniører kunne kontrollere denne adfærd, disse materialer kan bruges som kontakter, sensorer eller, eventuelt, grundlaget for nye elektroniske enheder. "Denne store ændring i ledningsevne for et materiale er den type ting, vi gerne vil kunne forudsige pålideligt, "Siger Kent.
Laboratorieforskere studerer også disse isolator-til-ledere med eksperimenter. Denne valideringsindsats giver tillid til forudsigelseskraften i deres beregningsmetoder i en række materialer. Teamet har bygget open-source software, kendt som QMCPACK, der nu er tilgængelig online og på alle beregningsfaciliteterne fra DOE Office of Science.
Kent og hans kolleger håber at kunne bygge op til højtemperatur-superledere og andre komplekse og mystiske materialer. Selvom forskere kender disse materialers brede egenskaber, Kent siger, "vi kan endnu ikke relatere dem til den faktiske struktur og elementerne i materialerne. Så det er en virkelig stor udfordring for det kondenserede fysikfelt."
De mest præcise kvantemekaniske modelleringsmetoder begrænser forskere til kun at undersøge nogle få atomer eller molekyler. Når forskere ønsker at studere større systemer, beregningsomkostningerne bliver hurtigt uhåndterlige. QMC tilbyder et kompromis:en beregnings størrelse stiger kubisk i forhold til antallet af elektroner, en mere overskuelig udfordring. QMC indeholder kun få kontrollerede tilnærmelser og kan anvendes på de mange atomer og elektroner, der er nødvendige. Den er velegnet til nutidens petascale supercomputere – i stand til en kvadrillion eller mere hvert sekund – og morgendagens exascale supercomputere, hvilket vil være mindst tusind gange hurtigere. Metoden kortlægger simuleringselementer relativt let på beregningsknuderne i disse systemer.
CPSFM-teamet fortsætter med at optimere QMCPACK til stadigt hurtigere supercomputere, herunder OLCF's Summit, som vil være fuldt operationel i januar 2019. Den højere hukommelseskapacitet på maskinens Nvidia Volta GPU'er - 16 gigabyte pr. grafikprocessorenhed sammenlignet med 6 gigabyte på Titan - øger allerede beregningshastigheden. Ved hjælp af OLCFs Ed D'Azevedo og Andreas Tillack, forskerne har implementeret forbedrede algoritmer, der kan fordoble hastigheden af deres større beregninger.
QMCPACK er en del af DOE's Exascale Computing Project, og teamet forventer allerede yderligere skaleringsudfordringer for at køre QMCPACK på fremtidige maskiner. For at udføre de ønskede simuleringer inden for ca. 12 timer på en exascale supercomputer, Kent vurderer, at de har brug for algoritmer, der er 30 gange mere skalerbare end dem i den nuværende version.
Selv med forbedret hardware og algoritmer, QMC -beregninger vil altid være dyre. Så Kent og hans team vil gerne bruge QMCPACK til at forstå, hvor billigere metoder går galt, så de kan forbedre dem. Derefter kan de gemme QMC -beregninger til de mest udfordrende problemer inden for materialevidenskab, Siger Kent. "Ideelt set lærer vi, hvad der får disse materialer til at være meget vanskelige at modellere og derefter forbedre billigere fremgangsmåder, så vi kan lave langt større scanninger af forskellige materialer."
Kombinationen af forbedrede QMC -metoder og en række beregningsmæssigt billigere modelleringsmetoder kan føre vejen til nye materialer og en forståelse af deres egenskaber. Det er dyrt at designe og teste nye forbindelser i laboratoriet, Siger Kent. Forskere kunne spare værdifuld tid og ressourcer, hvis de først kunne forudsige nye materialers opførsel i en simulering.
Plus, bemærker han, pålidelige beregningsmetoder kunne hjælpe forskere med at forstå egenskaber og processer, der er afhængige af individuelle atomer, der er ekstremt vanskelige at observere ved hjælp af eksperimenter. "Det er et sted, hvor der er stor interesse i at gå efter den grundlæggende videnskab, forudsige nye materialer og muliggøre teknologiske applikationer. "
 Varme artikler
Varme artikler
-
 Atomiske fætre slår sig sammen i en tidlig kvante -netværksknudeEn skematisk oversigt over tre noder i et kvantenetværk, forbundet med fiberoptiske kabler og forbundet til en central sensor. Kredit:V. Inlek/JQI og M. Lichtman/JQI Kvantecomputere i stor skala,
Atomiske fætre slår sig sammen i en tidlig kvante -netværksknudeEn skematisk oversigt over tre noder i et kvantenetværk, forbundet med fiberoptiske kabler og forbundet til en central sensor. Kredit:V. Inlek/JQI og M. Lichtman/JQI Kvantecomputere i stor skala, -
 Bedre integrerede kredsløb med glidesymmetriNonglide og glide symmetriske enhedsceller. Kredit:Xiao Tian Yan et al., DOI:10.1117/1.AP.3.2.026001 Surface plasmon polaritons (SPPer) er stærkt lokaliserede overfladebølger på grænsefladen melle
Bedre integrerede kredsløb med glidesymmetriNonglide og glide symmetriske enhedsceller. Kredit:Xiao Tian Yan et al., DOI:10.1117/1.AP.3.2.026001 Surface plasmon polaritons (SPPer) er stærkt lokaliserede overfladebølger på grænsefladen melle -
 De er der, og de er væk:ICARUS jagter en fjerde neutrinoICARUS – den største partikeldetektor i laboratoriets Short-Baseline Neutrino Program – og fylder den med 760 tons flydende argon, at flytte ICARUS tættere på drift og søgen efter en fjerde type neutr
De er der, og de er væk:ICARUS jagter en fjerde neutrinoICARUS – den største partikeldetektor i laboratoriets Short-Baseline Neutrino Program – og fylder den med 760 tons flydende argon, at flytte ICARUS tættere på drift og søgen efter en fjerde type neutr -
 Neutroner målt med en hidtil uset præcision ved hjælp af en magneto-gravitationsfældeUNCtau flaske fælden. En kombination af magnetfelter og tyngdekraft forhindrer neutroner i at undslippe beholderen. Kredit:Los Alamos National Laboratory En undersøgelse ledet af fysikere ved Indi
Neutroner målt med en hidtil uset præcision ved hjælp af en magneto-gravitationsfældeUNCtau flaske fælden. En kombination af magnetfelter og tyngdekraft forhindrer neutroner i at undslippe beholderen. Kredit:Los Alamos National Laboratory En undersøgelse ledet af fysikere ved Indi
- 15 af Spitzers største opdagelser fra 15 år i rummet
- Ny teknik til at udforske strukturelle dynamikker i nanoworld
- Håndhævelse af kønskvoter øger bestyrelseslokalernes mangfoldighed og kvalitet
- Hvordan hajer og andre dyr udviklede elektroreception for at finde deres bytte
- Bronzealderens minedriftssteder modtog leverancer af færdigforarbejdede fødevarer
- Er der en teknologisk løsning på akvatiske dødzoner?