


Nye fund kan føre til billigere solceller
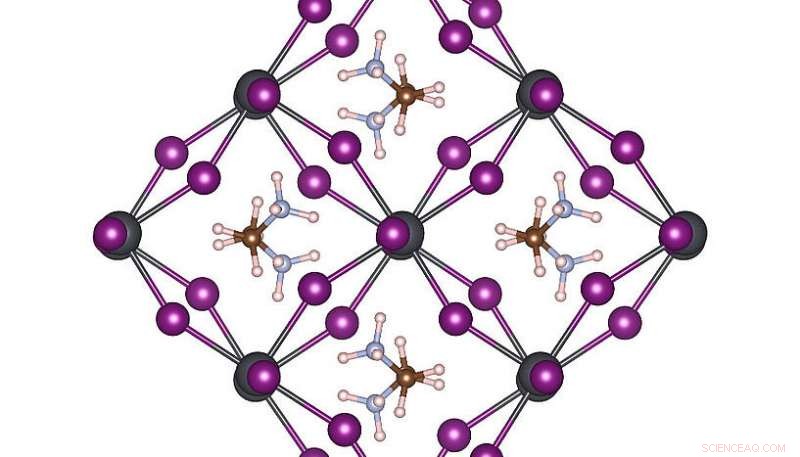
Atomstruktur med høj symmetri af MAPbI3 ved stuetemperatur. Kredit:Menno Bokdam/University of Vienna
På atomær skala kan materialer vise en rig palet af dynamisk adfærd, som direkte påvirker disse materialers fysiske egenskaber. I mange år, det har været en drøm at beskrive disse dynamikker i komplekse materialer ved forskellige temperaturer ved hjælp af computersimuleringer. Fysikere fra universitetet i Wien har udviklet en on-the-fly maskinlæringsmetode, der muliggør sådanne beregninger gennem direkte integration i den kvantemekanikbaserede Vienna Ab-initio Simulation Package (VASP). Selvlæringsmetodens alsidighed demonstreres af nye fund, offentliggjort i tidsskriftet Fysisk gennemgangsbreve , om faseovergangene af hybride perovskitter. Disse perovskitter er af stor videnskabelig interesse på grund af deres potentiale i solenergihøst og andre anvendelser.
Ved stuetemperatur, alle materialer bevæger sig konstant på atomskalaen. Selv fast sten består af atomer, der svinger rundt. Materialernes fysiske egenskaber er direkte forbundet med arrangementet af atomer i, såkaldte, krystalgitter. Afhængigt af temperaturen eller trykket kan dette arrangement ændre sig og derved påvirke materialets egenskaber. Man kan tænke på diamant, som er gennemsigtig og hård på grund af det periodiske arrangement af carbonatomer i diamantkrystallet. De samme atomer, arrangeret anderledes, resulterer i sort, sprød grafit. Det var allerede muligt nøjagtigt at beregne koordinaterne for atomerne i simple materialer ved forskellige temperaturer med kvantemekanisk molekylær dynamik (MD) simuleringer. Imidlertid, sådanne beregninger er beregningsmæssigt dyre og begrænser praktiske applikationer til et par hundrede atomer og begrænset simuleringstid.
Fysikere fra Computational Materials Physics-gruppen ved Universitetet i Wien har udviklet en ny tilgang, der overvinder disse begrænsninger og gør simuleringer af komplekse materialer til fremtidige energianvendelser mulige. Dette opnås ved at udvikle en effektiv og robust datadrevet selvlærende algoritme og, mest vigtigt, ved at integrere denne algoritme direkte i Wien Ab-initio Simulation Package (VASP). I den nye tilgang, "maskinen" kan hente, på egen hånd, de væsentlige ingredienser for en enklere modelbeskrivelse af de interagerende atomer under MD -simuleringer. Allerede efter beregning af et par hundrede tidstrin kan maskinen forudsige atomernes positioner præcist nok i det på hinanden følgende tidstrin. Maskinen er også i stand til at lave et skøn over dens nøjagtighed for de på hinanden følgende trin. Hvis fejlen er for høj, maskinen skifter gear og udfører den meget nøjagtige, men dyrt, MD -beregninger. Jo længere simuleringstiden går, jo mere maskinen lærer og jo mere præcis bliver den. På denne måde, færre og færre MD -beregninger er påkrævet, hvilket i sidste ende fører til den situation, hvor alle tidstrin foretages af maskinen. I øvrigt, den on-the-fly selvlæringsevne reducerer behovet for menneskelig indgriben, der kræves af andre eksisterende maskinlæringsmetoder.
For at demonstrere kraften i denne nye metode, forskerne har anvendt det til at studere overgangene mellem forskellige atomstrukturer i MAPbI 3 perovskit ved ændring af temperaturen. Dette materiale er meget populært på grund af dets potentiale som en ny billig solcellekomponent. Den er lavet af organiske molekyler, der hurtigt kan vende rundt, adskilt fra hinanden af et gitter bestående af bly- og iodidatomer. Afhængigt af temperaturen dannes tre forskellige krystalfaser. De atomare mekanismer nær overgangstemperaturen er meget vanskelige at bestemme ved eksperiment, og MD-simuleringer ville kræve mange års beregningstid selv på et moderne supercomputersystem. Efter at have lært, maskinen kan forudsige faseovergangstemperaturerne og gitterkonstanter for dette materiale med hidtil uset præcision. Den udviklede metode er generel og anvendelig for mange andre fremtidige materialevidenskabelige problemer og vil blive tilgængelig for forskere ordgående i den kommende version af VASP.
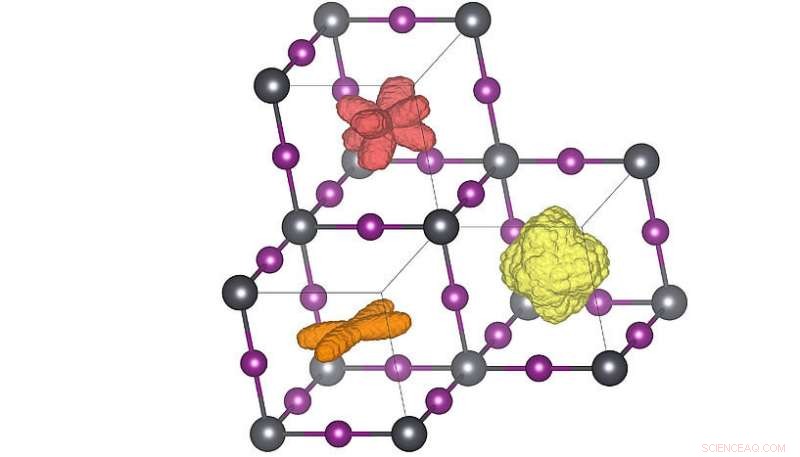
Tredimensionelle fordelinger af molekylets orientering i de tre forskellige krystalfaser. Når temperaturen hæves (orange→ rød→ gul) kan molekylerne opnå flere orienteringer. Den røde fordeling svarer til rumtemperaturstrukturen. Kredit:Menno Bokdam/University of Vienna
Sidste artikelMikroskopisk glasblæsning bruges til at lave små optiske linser
Næste artikelLøsning af et kondensmysterium
 Varme artikler
Varme artikler
-
 Sådan overlistes støj i kvantekommunikationSelv i tilstedeværelse af støj, kvanteinformationsoverførsel er mulig med et par særlige tricks. Kredit:IQOQI/Harald Ritsch Hvordan overfører man pålideligt kvanteinformation, når forbindelseskana
Sådan overlistes støj i kvantekommunikationSelv i tilstedeværelse af støj, kvanteinformationsoverførsel er mulig med et par særlige tricks. Kredit:IQOQI/Harald Ritsch Hvordan overfører man pålideligt kvanteinformation, når forbindelseskana -
 Forskere analyserer flokadfærd på buede overfladerStadige flokke på en kugle og en katenoid. Kredit:Suraj Shankar En mumlen af stære. Sætningen lyder som noget fra litteraturen eller titlen på en arthouse-film. Faktisk, det er beregnet til at b
Forskere analyserer flokadfærd på buede overfladerStadige flokke på en kugle og en katenoid. Kredit:Suraj Shankar En mumlen af stære. Sætningen lyder som noget fra litteraturen eller titlen på en arthouse-film. Faktisk, det er beregnet til at b -
 Måling af naturens loveNeutronforsøg ved ILL Grenoble. Kredit:Vienna University of Technology, TU Wien En fysisk konstant, som har stor betydning for grundforskningen, er nu blevet målt igen, med meget højere præcision
Måling af naturens loveNeutronforsøg ved ILL Grenoble. Kredit:Vienna University of Technology, TU Wien En fysisk konstant, som har stor betydning for grundforskningen, er nu blevet målt igen, med meget højere præcision -
 Integrering af nanokaviteter i optiske fibre med femtosekundlaserablationScanning elektronmikroskopi billede af den resulterende ensartethed og dimensioner af fotoniske krystaller induceret på nanofibre ved hjælp af femtosekund laser-induceret ablation. Kredit:University o
Integrering af nanokaviteter i optiske fibre med femtosekundlaserablationScanning elektronmikroskopi billede af den resulterende ensartethed og dimensioner af fotoniske krystaller induceret på nanofibre ved hjælp af femtosekund laser-induceret ablation. Kredit:University o
- Dødzonerapport afspejler forbedret vandkvalitet i Chesapeake Bay
- Solvinden, forklaret
- Hvordan forårsager mennesker erosion?
- Bioetiker diskuterer fire nøgler til at vide om muligheder, faldgruber ved genredigering
- Billede:Indgang til Hertz kammer
- Mansplaining:Nye løsninger på et trættende gammelt problem