


En ny metode til at studere polaroner i isolatorer og halvledere
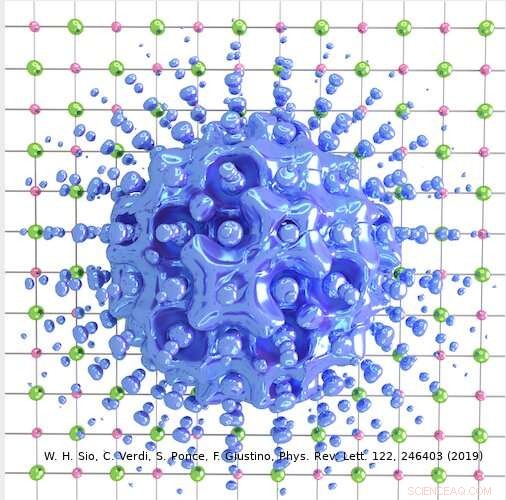
Kredit:Weng Hong Sio.
Et team af forskere ved University of Oxford har for nylig introduceret en ny måde at modellere polaroner på, en kvasipartikel, der typisk bruges af fysikere til at forstå interaktioner mellem elektroner og atomer i faste materialer. Deres metode, præsenteret i et papir offentliggjort i Fysisk gennemgangsbreve , kombinerer teoretisk modellering med beregningssimuleringer, muliggør dybdegående observationer af disse kvasipartikler i en lang række materialer.
I det væsentlige, en polaron er en sammensat partikel bestående af en elektron omgivet af en sky af fononer (dvs. gittervibrationer). Denne kvasipartikel er tungere end selve elektronen, og på grund af dens betydelige vægt kan den nogle gange blive fanget i et krystalgitter.
Polaroner bidrager til den elektriske strøm, der driver adskillige teknologiske værktøjer, inklusive organiske lysemitterende dioder og touchskærme. At forstå deres egenskaber er derfor af afgørende betydning, da det kunne være med til at udvikle den næste generation af forskellige enheder til belysning og optoelektronik.
"Tidligere arbejde med polaroner var baseret på idealiserede matematiske modeller, "Prof. Feliciano Giustino, lederen af det team, der udførte undersøgelsen, fortalte Phys.org. "Disse modeller har været meget nyttige til at forstå de grundlæggende egenskaber ved polaroner, men de tager ikke højde for strukturen af materialer på atomær skala, derfor er de ikke tilstrækkelige, når vi forsøger at studere virkelige materialer til praktiske anvendelser. Vores idé var at udvikle en beregningsmetode, der ville muliggøre systematiske undersøgelser af polaroner med forudsigelig nøjagtighed. "
Metoden udviklet af Giustinos team er baseret på densitet-funktionelle teori, som i øjeblikket er det mest populære værktøj til prædiktiv materialemodellering og design ved brug af kvantemekanik. En af de primære udfordringer, man støder på, når man studerer polaroner baseret på denne teori, er, at de nødvendige beregningsressourcer (CPU-timer) er proportionale med tredje potens af antallet af atomer, der skal simuleres. Med andre ord, hvis man studerede to krystaller med 10 og 20 atomer pr. enhedscelle, den beregning, der kræves for at studere den anden krystal, ville være 8 gange mere tidskrævende end den, der kræves for den første.
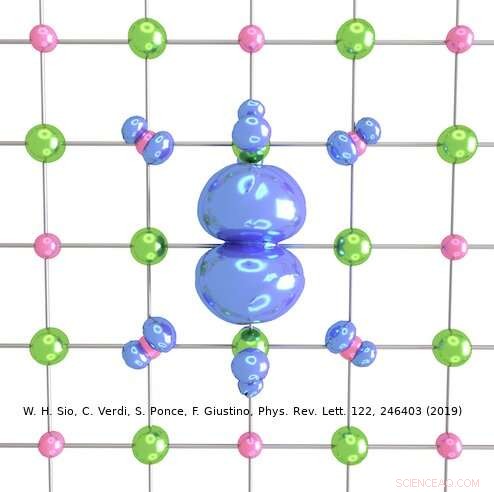
Kredit:Weng Hong Sio.
Da mange polaroner er 1-2 nanometer i størrelse, beregninger for at studere disse systemer ville kræve simuleringsceller med mindst 3, 000-5, 000 atomer. Men de nuværende beregningsmuligheder ville kæmpe for at opretholde sådanne simuleringer, og hver af de mange beregninger, der er nødvendige for at undersøge disse systemer, ville tage uger, selv når du bruger en moderne supercomputer.
"Vores idé var at forsøge at gøre denne proces mere effektiv ved at udnytte fremskridt inden for såkaldt tæthedsfunktionel forstyrrelsesteori, "Weng Hong Sio, den første forfatter til værket, forklaret. "Uden at gå ind på detaljerne, vi var i stand til at omforme problemet med at udføre en beregning af en polaron i en stor simuleringscelle til det enklere problem med at udføre flere beregninger i den mindste enhedscelle i krystallen. Denne strategi åbnede op for nye muligheder, som tidligere var utilgængelige."
Den tilgang, som Giustinos team har udtænkt, kan bruges til at beskrive både store og små polaroner. I deres undersøgelse, for eksempel, forskerne viste, hvordan det kan bruges til at beregne bølgefunktionerne, dannelsesenergier og spektral nedbrydning af polaroner i LiF og Li 2 O 2 forbindelser. Ved at bruge deres simuleringsmetode, de opdagede, at polaroner i simple salte og metaloxider, der bruges i batterier, har en langt rigere indre struktur end foreslået af tidligere værker på området.
"For eksempel, i det prototypiske salt lithiumfluorid, man troede tidligere, at polaronen stammer fra interaktionen mellem en elektron og longitidinale optiske fononer, dvs. gittervibrationerne, der er ansvarlige for krystallens dielektriske respons, " Sio forklarede. "Vi fandt ud af, at disse ikke er de eneste fononer involveret, og at interaktionen mellem elektronen og de piezoakustiske fononer (dvs. de vibrationer, der er ansvarlige for piezoelektricitet) også er vigtig."
Observationerne indsamlet af Giustinos hold ændrer det nuværende perspektiv på polaronerne i salt lithium fourid, hvilket er et meget simpelt system. Anvendelse af deres metode til mere komplekse systemer kunne afsløre endnu rigere strukturer, i sidste ende forbedre vores nuværende forståelse af deres egenskaber og informere udviklingen af nye materialer med skræddersyede polatroniske egenskaber. I deres fremtidige forskning, forskerne planlægger at bruge deres metode til at studere andre materialer, for yderligere at vurdere dens forudsigelsesevne og opnå en bedre forståelse af andre teknologisk vigtige materialer.
"Længere nede på linjen vil det være vigtigt at undersøge, hvad en polaron kan:for nu ved vi, at vi kan beregne den laveste energikonfiguration af et polaron, men vi har ingen anelse om, hvad der sker, hvis denne polaron udsættes for statiske elektriske eller magnetiske felter eller for elektromagnetisk stråling, " sagde Giustino. "Desuden, tætte interaktioner med eksperimentelle grupper vil være afgørende for at omsætte disse resultater til applikationer."
© 2019 Science X Network
Sidste artikelEnkelt smart glas afslører fremtiden for kunstigt syn
Næste artikelIndesluttet nanoskalalyd styrer lyset i en mikroresonator
 Varme artikler
Varme artikler
-
 Udforskning af topologi i biologiTop:Som et S, der bliver til et O, spektret af et biokemisk system kan gennemgå en topologisk overgang. Nederst til venstre:I topologisk beskyttet tilstand, det biokemiske system gennemgår kantcykluss
Udforskning af topologi i biologiTop:Som et S, der bliver til et O, spektret af et biokemisk system kan gennemgå en topologisk overgang. Nederst til venstre:I topologisk beskyttet tilstand, det biokemiske system gennemgår kantcykluss -
 Forskere regner sig frem til renere kulværkerVed hjælp af HLRS Hazel Hen -maskinen, RWTH Aachen University forskere var i stand til at køre en DNS -simulering på et system på 45, 000 partikler i Kolmogorov -skalaen. Så vidt teamet ved, dette er
Forskere regner sig frem til renere kulværkerVed hjælp af HLRS Hazel Hen -maskinen, RWTH Aachen University forskere var i stand til at køre en DNS -simulering på et system på 45, 000 partikler i Kolmogorov -skalaen. Så vidt teamet ved, dette er -
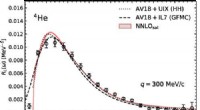 Atomkerner og leptoner:Milepæl nået i beregningen af tværsnitFigur 1. Longitudinel responsfunktion for 4 Han kl q =300 MeV/c. HH resultater taget fra Ref. [44], GFMC resultater fra Ref. [43], og eksperimentelle data fra Ref. [45]. Kredit:DOI:10.1103/PhysRev
Atomkerner og leptoner:Milepæl nået i beregningen af tværsnitFigur 1. Longitudinel responsfunktion for 4 Han kl q =300 MeV/c. HH resultater taget fra Ref. [44], GFMC resultater fra Ref. [43], og eksperimentelle data fra Ref. [45]. Kredit:DOI:10.1103/PhysRev -
 Indesluttet nanoskalalyd styrer lyset i en mikroresonatorScanningelektronmikrofotografi af den nye Brillouin-spredningsmikrosøjleanordning. Diameteren er 4,5 mikrometer. De falske farver markerer den indre del, der begrænser højfrekvente vibrationer (i oran
Indesluttet nanoskalalyd styrer lyset i en mikroresonatorScanningelektronmikrofotografi af den nye Brillouin-spredningsmikrosøjleanordning. Diameteren er 4,5 mikrometer. De falske farver markerer den indre del, der begrænser højfrekvente vibrationer (i oran
- Undersøgelse af den supergigantiske stjerne Betelgeuse afslører årsagen til dens pulseringer
- Edward Adelbert Doisy
- Qualcomm tilbagekøber 16 milliarder dollar af sine aktier fra banker
- Forskere skaber 3D-printede, mikroskopiske gassensorer:Malerpaller uden et strejf af pigment
- Chinas Didi øger udfordringen til Uber med Australien -skub
- Opdræt af laks