


Teori afslører arten af defekter i siliciumcarbidkrystaller
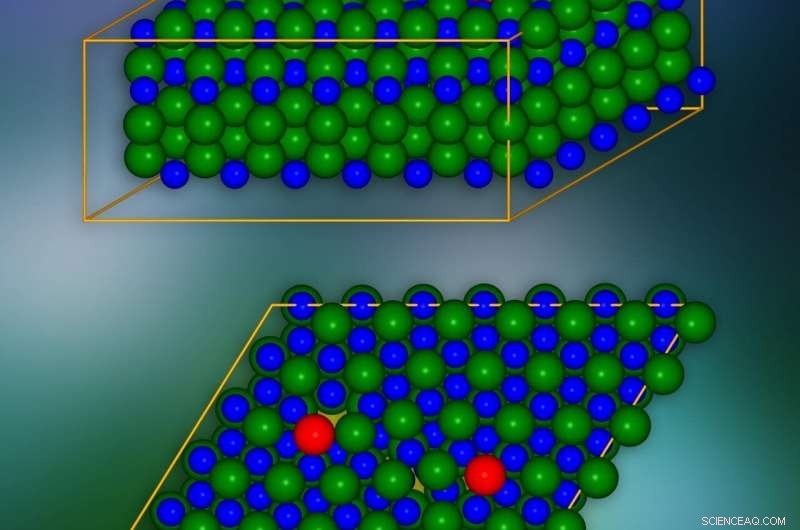
Siliciumcarbid krystalmodel med kantforskydninger indført på steder markeret med rødt. Et enkelt krystallografisk plan er præsenteret i bunden. De steder, hvor elektriske ladninger kan 'lække' til nabolag, er markeret med gult. Kredit:IFJ PAN
Ufuldkommenheder i krystalstruktur, især kantforskydninger af langstrakt karakter, dybt ændre grundlæggende egenskaber af hele materialet og, som konsekvens, begrænse dets anvendelser drastisk. Ved at bruge siliciumcarbid som eksempel, fysikere fra Krakow og Warszawa har vist, at selv sådanne beregningskrævende defekter med held kan undersøges med atomnøjagtighed ved hjælp af en smart konstrueret, lille i størrelsen, model.
Matematik elsker perfektion. Desværre, perfektion elsker ikke fysisk virkelighed. Teoretikere, der modellerer krystaller, har længe forsøgt at inkludere defekter i rigtige krystallinske strukturer og forudsige deres indvirkning på materialers fysiske egenskaber. modellerne, baseret på resultaterne af forskellige eksperimenter, har beskrevet ændringer i et materiales grundlæggende egenskaber uden at forklare de virkelige årsager og virkninger af de forekommende fænomener.
En ny model af siliciumcarbid (SiC), bygget af fysikere fra Institute of Nuclear Physics ved det polske videnskabsakademi (IFJ PAN) i Krakow, har givet dem mulighed for at demonstrere, at det nu er muligt at studere krystaller ab initio med så komplekse defekter som kantforskydninger og at forklare deres karakteristika ved processer, der foregår på atomær skala. Dette spektakulære resultat, for nylig præsenteret på konferencen Multiscale Phenomena in Molecular Matter 2019 i Krakow, blev opnået af IFJ PAN-fysikere i samarbejde med Institute of Fundamental Technological Research ved det polske videnskabsakademi og instituttet for højtryksfysik ved det polske videnskabsakademi, begge placeret i Warszawa.
"Vi forsøgte at finde de mekanismer, der på atomniveau er ansvarlige for at sænke nedbrydningsspændingen i siliciumcarbidkrystaller. Vores ab initio-beregninger fører til en kvalitativ forståelse af problemet og bidrager til at forklare detaljerne i dette fænomen, " siger Dr. Jan Lazewski, professor ved IFJ PAN.
Ab initio-beregninger har nu en lang historie relateret til Nobelprisen til Walter Kohn og John Pople i 1998 (men til lineære krystaldefektsimuleringer er de først for nylig blevet introduceret). Dette udtryk bruges til at beskrive beregninger udført ved hjælp af kvantemekaniske ligninger, kun understøttet af viden om atomets struktur og krystallers symmetri. Der er ingen direkte information fra eksperimenter i sådanne modeller, hvilket betyder, at de også kan bruges til at analysere materialer, der aldrig er blevet undersøgt eller endda syntetiseret før. På grund af relativt betydelig komplikation af problemet, indtil videre har ab initio-beregninger fungeret, højst, i tilfælde af punktfejl, relateret til vakanser (manglende atomer eller huller i krystalstrukturen) samt tilsætningsstoffer indført i krystallen.
Det var ikke uden grund, at Krakow-forskerne brugte siliciumcarbid. Egenskaberne ved denne halvleder er så interessante, at den tidligere endda blev betragtet som en efterfølger til silicium. Dens båndgab (barrieren ladningen skal overvinde for at komme fra valensbåndet til ledningsbåndet og lede strøm) er næsten tre gange større end i silicium, den tilladte ledningsstrømtæthed - dobbelt så stor, evnen til at sprede varme - mere end tre gange større, og afskæringsfrekvensen for krystaldrift så mange som seks gange større. Ud over, siliciumcarbidsystemer kan fungere ved temperaturer op til 650 grader Celsius, mens siliciumsystemer allerede begynder at få problemer ved 120 grader Celsius. SiC har også et højt smeltepunkt, det er svært, modstandsdygtig over for syre og stråling. Dens ulemper omfatter frem for alt prisen:mens to-tommers siliciumwafers kun koster et par dollars, værdien af lignende siliciumcarbidskiver løber op i tusindvis. Siliciumcarbidkrystaller af lav kvalitet er et populært slibende materiale, også brugt i skudsikre veste og i bremseskiverne på verdens dyreste biler, såsom Lamborghini eller Bugatti. Højkvalitetskrystaller bruges til at fremstille spejle til teleskoper og i højspændingsenheder med høj temperaturbestandighed.
På atomniveau, siliciumcarbidkrystaller er sammensat af mange flade lag, der er anbragt oven på hinanden. Hvert lag ligner en honeycomb:det består af sekskantede celler, hvor siliciumcarbidmolekylerne er placeret lodret i hjørnerne. Hvert to tilstødende lag kan kombineres på tre måder. Flerlags 'sandwichene' med forskellige layouts skaber såkaldte polytyper, hvoraf der findes mere end 250 for siliciumcarbid. Gruppen fra IFJ PAN brugte 4H-SiC polymorfen.
"Når man modellerer sådanne strukturer, et af hovedproblemerne er beregningsmæssig kompleksitet. En model af ren krystal, fri for blandinger eller dislokationer, er kendetegnet ved høj symmetri og kan beregnes selv på få minutter. For at udføre en beregning for et materiale med dislokation, vi har brug for måneder med at arbejde på en højtydende computer, " understreger Dr. Pawel Jochym, professor ved IFJ PAN.
Problemerne med kantdislokationer skyldes omfanget af deres indflydelse på materialets krystalstruktur. Som en illustration, de kan sammenlignes med problemet med at skjule et hul i en række af fliser på et gulv. Mellemrummet kan "camoufleres" ved at flytte fliserne på tilstødende rækker, men fejlen vil altid forblive synlig. Kantforskydninger som følge af manglen på hele længder eller områder af atomer/molekyler i individuelle krystallag virker på samme måde, påvirker positionerne af atomer og molekyler i mange tilstødende lag. Og da dislokationerne kan strække sig over lange afstande, i praksis omfatter forstyrrelserne forårsaget af dem hele krystallen.
De mest interessante fænomener finder sted i dislokationskernen, dvs. i nærheden af kanten af det beskadigede lag af krystalnetværket. For at eliminere langtrækkende effekter forårsaget af en enkelt dislokation, og dermed reducere antallet af atomer, der overvejes, markant, et trick blev brugt:en anden dislokation af den modsatte effekt blev indført. På denne måde virkningen af den første dislokation over længere afstande blev kompenseret for.
SiC-krystalmodellen bestod af omkring 400 atomer. Simuleringerne viste, at i lagene af krystaller, langs kanten af defektens kerne, 'tunneler' optræder i form af kanaler med reduceret ladningstæthed. De sænker potentialbarrieren lokalt og får elektriske ladninger til at 'lække' fra valensbåndet. Ud over, i det forbudte hul, som i isolatoren garanterer mangel på elektrisk ledningsevne, der opstår forhold, som reducerer dens bredde og effektivitet til at begrænse ladningsstrømmen. Det blev vist, at disse tilstande stammer fra atomer placeret i dislokationskernen.
"Situationen kan sammenlignes med en dyb, stejle kløft, som et egern forsøger at krydse. Hvis bunden af kløften er tom, egernet kommer ikke til den anden side. Imidlertid, hvis der er et antal træer i bunden, der er høje nok, egernet kan hoppe over deres toppe til den anden side af kløften. I den krystal vi modellerede, egerne er de elektriske ladninger, valensbåndet er den ene kant af kløften, ledningsbåndet er det andet, og træerne er de førnævnte tilstande forbundet med atomerne i dislokationskernen, " siger prof. Lazewski.
Nu hvor de mekanismer, der er ansvarlige for at sænke tærsklen for energibarrieren, er blevet kendt på atomniveau, der er store muligheder for at eksperimentere. Den foreslåede mekanisme skal verificeres for at kunne bruge den til at begrænse den negative indflydelse af de testede defekter. Heldigvis, der er allerede tekniske muligheder for dette.
"Fremtiden vil bekræfte, om vores ideer vil blive bekræftet i deres helhed. vi er sikre på vores models skæbne og den præsenterede tilgang til simulering af kantforskydninger. Vi ved allerede, at ab initio-modellen har bevist sit værd i konfrontation med visse eksperimentelle data, " slutter prof. Jochym.
Sidste artikelKunsten at orme gennem trange rum
Næste artikelEn ny måde at måle, hvordan vand bevæger sig
 Varme artikler
Varme artikler
-
 Tre kendte forskere:Heusler, Weyl og BerryFiguren viser forbindelsen mellem Weyl fermioner og Berry -fasen og dens realisering i Heusler -familien af forbindelser. I det øverste panel, vi præsenterer et typisk atomarrangement af en hel-Heus
Tre kendte forskere:Heusler, Weyl og BerryFiguren viser forbindelsen mellem Weyl fermioner og Berry -fasen og dens realisering i Heusler -familien af forbindelser. I det øverste panel, vi præsenterer et typisk atomarrangement af en hel-Heus -
 Metal Organic Framework (MOF) mikrokrystaller til flerfarvet bredbåndslasingSkematisk over enakset homøpitaksial vækst af ZJU-68 krystaller. Den organiske brodannende ligand H2CPQC giver en højere chelateringsstedtæthed langs krystalaksens retning, og den signifikante forskel
Metal Organic Framework (MOF) mikrokrystaller til flerfarvet bredbåndslasingSkematisk over enakset homøpitaksial vækst af ZJU-68 krystaller. Den organiske brodannende ligand H2CPQC giver en højere chelateringsstedtæthed langs krystalaksens retning, og den signifikante forskel -
 Sådan oprettes en fuld adder med MultiMedia LogicMultiMedia Logic er et gratis program til at lære at designe boolske kredsløb, såsom multiplexere, halv adders og fulde adders. Logiske tilføjere udfører binær tilføjelse på to vilkårligt store base-t
Sådan oprettes en fuld adder med MultiMedia LogicMultiMedia Logic er et gratis program til at lære at designe boolske kredsløb, såsom multiplexere, halv adders og fulde adders. Logiske tilføjere udfører binær tilføjelse på to vilkårligt store base-t -
 Genbesøger Clebschs tidlige artikler om strømmen af inkompressible væskerKredit:CC0 Public Domain Ny analyse af to nyligt oversatte artikler, første gang udgivet i 1850erne, vurderer de tidlige metoder brugt af Alfred Clebsch til at beskrive strømmen af inkompressibl
Genbesøger Clebschs tidlige artikler om strømmen af inkompressible væskerKredit:CC0 Public Domain Ny analyse af to nyligt oversatte artikler, første gang udgivet i 1850erne, vurderer de tidlige metoder brugt af Alfred Clebsch til at beskrive strømmen af inkompressibl
- Ændring af elevernes holdning til matematik forbedrer testresultaterne
- Fakta om solen til tredje grader niveau
- Astrofysikere forbinder oplysning af pulsarvindtågen til pulsars spin-down-hastighedsovergang
- De positive effekter af genteknologi
- Familier fjernundervisningsoplevelse under lockdown mere positiv end almindeligt antaget
- Italienske astronomer opdager en ny stjernehob