


Biologiske mønsterdannende systemer karakteriserede bedre gennem geometri end simuleringer

Ligesom fugleflokkers kollektive bevægelser, mønstrene skyldes de samordnede interaktioner mellem mange individuelle partikler uden en central koordinator. Kredit:CC0 Public Domain
Ludwig-Maximilians-Universitaet (LMU) i München har fysikere introduceret en ny metode, der gør det muligt systematisk at karakterisere biologiske mønsterdannende systemer ved hjælp af matematisk analyse. Tricket ligger i brugen af geometri til at karakterisere dynamikken.
Mange vitale processer, der finder sted i biologiske celler, afhænger af dannelsen af selvorganiserende molekylære mønstre. For eksempel, definerede rumlige fordelinger af specifikke proteiner regulerer celledeling, cellemigration og cellevækst. Disse mønstre skyldes de samordnede interaktioner mellem mange individuelle makromolekyler. Ligesom fugleflokkers kollektive bevægelser, disse processer behøver ikke en central koordinator. Hidtil, matematisk modellering af dannelse af proteinmønstre i celler er i vid udstrækning blevet udført ved hjælp af detaljerede computerbaserede simuleringer. Nu, LMU -fysikere under ledelse af professor Erwin Frey rapporterer om udviklingen af en ny metode, der giver mulighed for en systematisk matematisk analyse af mønsterdannelsesprocesser, og afdækker deres underliggende fysiske principper. Den nye tilgang beskrives og valideres i et papir, der vises i journalen Fysisk gennemgang X .
Undersøgelsen fokuserer på det, der kaldes 'massekonserverende' systemer, hvor interaktionerne påvirker tilstanden af de involverede partikler, men ændr ikke det samlede antal partikler, der er til stede i systemet. Denne betingelse er opfyldt i systemer, hvor proteiner kan skifte mellem forskellige konformationelle tilstande, der tillader dem at binde til en cellemembran eller til at danne forskellige multikomponentkomplekser, for eksempel. På grund af kompleksiteten af den ikke -lineære dynamik i disse systemer, mønsterdannelse er hidtil blevet undersøgt ved hjælp af tidskrævende numeriske simuleringer. "Nu kan vi forstå de markante træk ved mønsterdannelse uafhængigt af simuleringer ved hjælp af enkle beregninger og geometriske konstruktioner, "forklarer Fridtjof Brauns, hovedforfatter til det nye papir. "Den teori, vi præsenterer i denne rapport, giver i det væsentlige en bro mellem de matematiske modeller og den kollektive adfærd for systemets komponenter."
Den centrale indsigt, der førte til teorien, var erkendelsen af, at ændringer i den lokale antal tæthed af partikler også vil ændre positionerne for lokale kemiske ligevægte. Disse skift genererer igen koncentrationsgradienter, der driver partiklernes diffusive bevægelser. Forfatterne fanger dette dynamiske samspil ved hjælp af geometriske strukturer, der kendetegner den globale dynamik i et multidimensionelt faserum. "Systemers kollektive egenskaber kan direkte afledes af de topologiske forhold mellem disse geometriske konstruktioner, fordi disse objekter har konkrete fysiske betydninger - som repræsentationer af banerne for skiftende kemiske ligevægte, for eksempel.
"Det er grunden til, at vores geometriske beskrivelse giver os mulighed for at forstå, hvorfor de mønstre, vi observerer i celler, opstår. Med andre ord, de afslører de fysiske mekanismer, der bestemmer samspillet mellem de involverede molekylære arter, "siger Frey." Desuden de grundlæggende elementer i vores teori kan generaliseres til at håndtere en lang række systemer, hvilket igen baner vejen til en omfattende teoretisk ramme for selvorganiserende systemer. "
 Varme artikler
Varme artikler
-
 Ny plan for mere stabile kvantecomputereManuel Grimm er teoretisk fysiker ved Paul Scherrer Instituttet og arbejder på grundlaget for at bygge fremtidige kvantecomputere. Kredit:Paul Scherrer Institute/Markus Fischer Forskere ved Paul S
Ny plan for mere stabile kvantecomputereManuel Grimm er teoretisk fysiker ved Paul Scherrer Instituttet og arbejder på grundlaget for at bygge fremtidige kvantecomputere. Kredit:Paul Scherrer Institute/Markus Fischer Forskere ved Paul S -
 Observation af ikke-triviel superledning på overfladen af type II Weyl semimetalKredit:CC0 Public Domain Topologiske superledere, med superledende hul i bulk og Majorana fermiontilstande på overfladen eller kanten, er et af de mest eftertragtede kvantematerialer. Topologisk s
Observation af ikke-triviel superledning på overfladen af type II Weyl semimetalKredit:CC0 Public Domain Topologiske superledere, med superledende hul i bulk og Majorana fermiontilstande på overfladen eller kanten, er et af de mest eftertragtede kvantematerialer. Topologisk s -
 Metamaterialer bøjer bølger af alle slagsEn fordybet overflade med cylindre som overfladen af en Lego-mursten danner et ikke-metallisk ledende materiale. Metamaterialet absorberer elektromagnetisk energi uden opvarmning. Kredit:Duke Univer
Metamaterialer bøjer bølger af alle slagsEn fordybet overflade med cylindre som overfladen af en Lego-mursten danner et ikke-metallisk ledende materiale. Metamaterialet absorberer elektromagnetisk energi uden opvarmning. Kredit:Duke Univer -
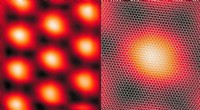 Ligheder i de isolerende tilstande af snoet to -lags grafen og kopaterHøjopløsnings scanning tunneling mikroskop (STM) billede af snoet to -lags grafen i den magiske vinkel, hvor elektroninteraktioner maksimeres. Højre:En zoom ind på STM -billedet med de tilsvarende git
Ligheder i de isolerende tilstande af snoet to -lags grafen og kopaterHøjopløsnings scanning tunneling mikroskop (STM) billede af snoet to -lags grafen i den magiske vinkel, hvor elektroninteraktioner maksimeres. Højre:En zoom ind på STM -billedet med de tilsvarende git
- Sådan beregnes termoelementfølsomhed
- Røntgenstråler kan være en bedre måde at kommunikere i rummet på
- Den oprindelige befolkning på det gamle Sicilien var aktive handlende
- Undersøgelse afslører skoleopsparingskonti kan tørre ud i finansielle ørkener
- Hvorfor folk opholder sig i katastrofeudsatte byer
- Fakta om skifer Rock