


Ny effektiv kvantealgoritme overgår Quantum Phase Estimation-normen
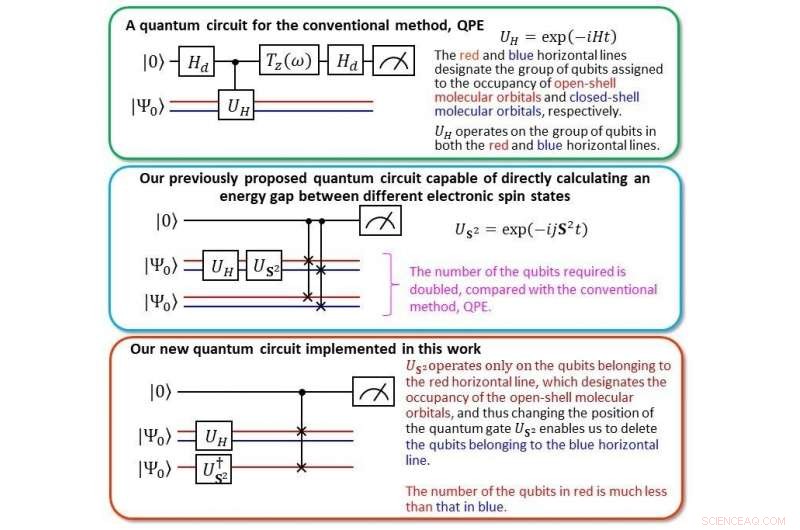
Sammenligning af det nye kvantekredsløb med vores tidligere Kredit:Kenji Sugisaki, Takeji Takui, Kazunobu Sato
Kvantecomputere har set meget opmærksomhed på det seneste, da de forventes at løse visse problemer, der ligger uden for normale computeres muligheder. Det primære for disse problemer er at bestemme de elektroniske tilstande af atomer og molekyler, så de kan bruges mere effektivt i en række forskellige industrier - fra design af lithium-ion-batterier til i silico-teknologier i lægemiddeludvikling. En almindelig måde, videnskabsmænd har nærmet sig dette problem på, er ved at beregne de samlede energier af de individuelle tilstande af et molekyle eller atom og derefter bestemme forskellen i energi mellem disse tilstande. I naturen, mange molekyler vokser i størrelse og kompleksitet, og omkostningerne til at beregne denne konstante flux er uden for enhver traditionel computers evne eller i øjeblikket etablere kvantealgoritmer. Derfor, teoretiske forudsigelser af de samlede energier har kun været mulige, hvis molekyler ikke er store og isolerede fra deres naturlige miljø.
"For at kvantecomputere skal være en realitet, dens algoritmer skal være robuste nok til nøjagtigt at forudsige de elektroniske tilstande af atomer og molekyler, som de findes i naturen, " oplyser Kenji Sugisaki og Takeji Takui fra Graduate School of Science, Osaka City University.
I december 2020, Sugisaki og Takui, sammen med deres kolleger, førte et team af forskere til at udvikle en kvantealgoritme, de kalder Bayesian eXchange koblingsparameterberegner med Broken-symmetri wave-funktioner (BxB), der forudsiger atomers og molekylers elektroniske tilstande ved direkte at beregne energiforskellene. De bemærkede, at energiforskelle i atomer og molekyler forbliver konstante, uanset hvor komplekse og store de bliver på trods af deres samlede energier vokser som systemets størrelse. "Med BxB, vi undgik den almindelige praksis med at beregne de samlede energier og målrettede energiforskellene direkte, holde beregningsomkostningerne inden for polynomisk tid, " siger de. "Siden da, vores mål har været at forbedre effektiviteten af vores BxB-software, så den kan forudsige den elektroniske tilstand af atomer og molekyler med kemisk præcision."
Ved at bruge beregningsomkostningerne for en velkendt algoritme kaldet Quantum Phase Estimation (QPE) som benchmark, "Vi beregnede de vertikale ioniseringsenergier af små molekyler som CO, O 2 , CN, F 2 , H 2 Åh, NH 3 inden for 0,1 elektronvolt (eV) præcision, " siger holdet, ved at bruge halvdelen af antallet af qubits, bringer beregningsomkostningerne på niveau med QPE.
Deres resultater vil blive offentliggjort online i marts-udgaven af Journal of Physical Chemistry Letters .
Ioniseringsenergi er en af de mest fundamentale fysiske egenskaber ved atomer og molekyler og en vigtig indikator for at forstå styrken og egenskaberne af kemiske bindinger og reaktioner. Kort sagt, nøjagtigt at forudsige ioniseringsenergien giver os mulighed for at bruge kemikalier ud over den nuværende norm. I fortiden, det var nødvendigt at beregne energierne i de neutrale og ioniserede tilstande, men med BxB kvantealgoritmen, ioniseringsenergien kan opnås i en enkelt beregning uden at inspicere de individuelle totale energier i neutral og ioniseret tilstand. "Fra numeriske simuleringer af det kvantelogiske kredsløb i BxB, vi fandt ud af, at beregningsomkostningerne for at udlæse ioniseringsenergien er konstant uanset atomnummeret eller molekylets størrelse, " siger holdet, "og at ioniseringsenergien kan opnås med en høj nøjagtighed på 0,1 eV efter at have ændret længden af det kvantelogiske kredsløb til at være mindre end en tiendedel af QPE."
Med udviklingen af kvantecomputerhardware, Sugisaki og Takui, sammen med deres team, forventer, at BxB kvantealgoritmen udfører højpræcise energiberegninger for store molekyler, der ikke kan behandles i realtid med konventionelle computere.
 Varme artikler
Varme artikler
-
 NASA ønsker at skabe det sejeste sted i universetKunstnerens koncept om en atomchip til brug ved NASAs Cold Atom Laboratory (CAL) ombord på den internationale rumstation. CAL vil bruge lasere til at afkøle atomer til ultrakølede temperaturer. Kredit
NASA ønsker at skabe det sejeste sted i universetKunstnerens koncept om en atomchip til brug ved NASAs Cold Atom Laboratory (CAL) ombord på den internationale rumstation. CAL vil bruge lasere til at afkøle atomer til ultrakølede temperaturer. Kredit -
 Forskere annoncerer foton-fonon-gennembrudTopologisk distinkte fotoniske krystaller (orange og blå) med et lag af hexagonalt bornitrid på toppen muliggør kobling af topologisk lys og gittervibrationer for at danne chirale halvlys-halvvibratio
Forskere annoncerer foton-fonon-gennembrudTopologisk distinkte fotoniske krystaller (orange og blå) med et lag af hexagonalt bornitrid på toppen muliggør kobling af topologisk lys og gittervibrationer for at danne chirale halvlys-halvvibratio -
 Forskellen mellem enkle og sammensatte maskinerI den generelle forstand er en maskine et apparat, der bruger energi til at udføre arbejde. Maskiner har et enormt udvalg af applikationer inden for det industrielle, kommercielle, private og ethve
Forskellen mellem enkle og sammensatte maskinerI den generelle forstand er en maskine et apparat, der bruger energi til at udføre arbejde. Maskiner har et enormt udvalg af applikationer inden for det industrielle, kommercielle, private og ethve -
 Spiser kejserpingviner nok? Forskere måler fouragering succes ved at spionere med time-lapse videoI en ny undersøgelse, videnskabsmænd siger, at de kan overvåge pingvinkoloniernes helbred - som denne ved Pointe Géologie i Adélie Land i Antarktis - ved at oprette langtidsobservatorier, der viderese
Spiser kejserpingviner nok? Forskere måler fouragering succes ved at spionere med time-lapse videoI en ny undersøgelse, videnskabsmænd siger, at de kan overvåge pingvinkoloniernes helbred - som denne ved Pointe Géologie i Adélie Land i Antarktis - ved at oprette langtidsobservatorier, der viderese
- Microsoft tilbyder softwareværktøjer til at sikre valg
- Acetone plus lys skaber et grønt jetbrændstoftilsætningsstof
- Hvilke egenskaber har vulkaner?
- Ammoniakrigt hagl kaster nyt lys over Jupiters vejr
- Hvorfor er videnskabsmænd så begejstrede for en nylig hævdet kvantecomputermilepæl?
- Forskning omdefinerer den nedre grænse for planetstørrelses beboelighed