


Ny bayesiansk kvantealgoritme beregner direkte energiforskellen for et atom og et molekyle
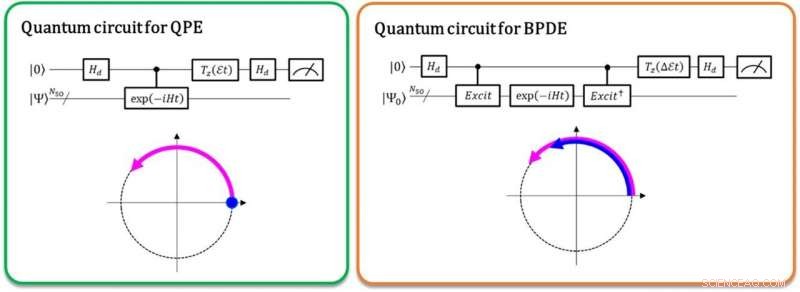
Til venstre:Faseforskellen mellem | 0⟩ | Ψ⟩ og exp (-iEt) | 1⟩ | Ψ⟩ giver den samlede energi E. Den buede pil i lilla angiver faseudviklingen af | Ψ⟩ i tid. Til højre:Faseforskellen mellem exp (-iE0t) | 0⟩ | Ψ0⟩ og exp (-iE1t) | 1⟩ | Ψ1⟩ giver energiforskellen E1-E0, direkte. De buede pile i blå og lilla angiver faseudviklingen af | Ψ0⟩ og | Ψ1⟩, henholdsvis. Kredit:K. Sugisaki, K. Sato og T. Takui
Som nyligt rapporteret af tidsskriftet Fysisk kemi Kemisk fysik , forskere fra Graduate School of Science ved Osaka City University har udviklet en kvantealgoritme, der kan forstå de elektroniske tilstande i atom- eller molekylære systemer ved direkte at beregne energiforskellen i deres relevante tilstande. Implementeret som en bayesisk fase anderledes estimering, algoritmen bryder fra konventionen ved ikke at fokusere på forskellen i samlede energier beregnet ud fra udviklingen før og efter fase, men ved at følge udviklingen af selve energiforskellen.
"Næsten alle kemiproblemer diskuterer energiforskellen, ikke molekylens samlede energi selv "siger forskningsleder og specielt udpeget foredragsholder Kenji Sugisaki, "også, molekyler med tunge atomer, der vises i den nedre del af det periodiske system, har store samlede energier, men størrelsen af energiforskellen diskuteret i kemi, såsom elektroniske excitationstilstande og ioniseringsenergier, afhænger ikke meget af molekylets størrelse. "Denne idé førte Sugisaki og hans team til at implementere en kvantealgoritme, der direkte beregner energiforskelle i stedet for samlede energier, skabe en fremtid, hvor skalerbare eller praktiske kvantecomputere sætter os i stand til at udføre egentlig kemisk forskning og materialeudvikling.
I øjeblikket, kvantecomputere er i stand til at udføre de fulde konfigurationsinteraktionsberegninger (fuld-CI), der giver optimale molekylære energier med en kvantealgoritme kaldet kvantefasestimering (QPE), bemærker, at fuld-CI-beregningen for betydelige molekylære systemer er umulig at håndtere med alle supercomputere. QPE er afhængig af, at en bølgefunktion, | Ψ⟩ som angiver den matematiske beskrivelse af et mikroskopisk systems kvantetilstand-i dette tilfælde den matematiske løsning af Schrödinger-ligningen for det mikroskopiske system, såsom et atom eller et molekyle-ændrer tids-evolutionært sin fase afhængigt af dets samlede energi. I den konventionelle QPE, kvantesuperpositionstilstanden (| 0⟩ | Ψ⟩+| 1⟩ | Ψ⟩) ⁄ √2 er forberedt, og indførelsen af en kontrolleret tidsudviklingsoperator får | Ψ⟩ kun til at udvikle sig i tide, når den første qubit angiver tilstanden | 1⟩. Dermed, tilstanden | 1⟩ skaber en kvantefase af efterudviklingen i tid, mens | 0⟩ angiver den for præ-evolutionens. Faseforskellen mellem for- og efterudviklingen giver systemets samlede energi.
Forskerne ved Osaka City University generaliserer den konventionelle QPE til den direkte beregning af forskellen i den samlede energi mellem to relevante kvantetilstande. I den nyligt implementerede kvantealgoritme kaldet Bayesian phase difference estimation (BPDE), superpositionen af de to bølgefunktioner, (| 0⟩ | Ψ 0 ⟩ + | 1⟩ | Ψ 1 ⟩) ⁄ √2, hvor | Ψ 0 ⟩ Og | Ψ 1 ⟩ Betegner den bølgefunktion, der er relevant for hver tilstand, henholdsvis, er forberedt, og forskellen i fasen mellem | Ψ 0 ⟩ Og | Ψ 1 ⟩ Efter den tid, evolution af superpositionen giver direkte forskellen i den totale energi mellem de to involverede bølgefunktioner. "Vi understreger, at algoritmen følger udviklingen af energiforskellen over tid, som er mindre tilbøjelig til støj end individuelt at beregne den samlede energi af et atom eller molekyle. Dermed, algoritmen passer til behovet for kemiproblemer, der kræver præcis energienøjagtighed. "fastslår forskningsvejleder og professor emeritus Takeji Takui.
Tidligere har denne forskergruppe udviklede en kvantealgoritme, der direkte beregner energiforskellen mellem elektroniske tilstande (spin -tilstande) med forskellige spin -kvantetal (K. Sugisaki, K. Toyota, K. Sato, D. Shiomi, T. Takui, Chem. Sci. 2021, 12 , 2121–2132.). Denne algoritme, imidlertid, kræver flere qubits end den konventionelle QPE og kan ikke anvendes på beregningen af energiforskel mellem de elektroniske tilstande med lige store spin -kvantetal, hvilket er vigtigt for spektral tildeling af UV-synlige absorptionsspektre. BPDE -algoritmen udviklet i undersøgelsen overvinder disse spørgsmål, hvilket gør det til en meget alsidig kvantealgoritme.
 Varme artikler
Varme artikler
-
 Design af lasere baseret på kvantefysikGermán J. de Valcárcel Gonzalvo. Kredit:Asociacion RUVID Et forskerhold på fem lande koordineret af Germán J. de Valcárcel Gonzalvo, professor i optik ved universitetet i Valencia, har udviklet en
Design af lasere baseret på kvantefysikGermán J. de Valcárcel Gonzalvo. Kredit:Asociacion RUVID Et forskerhold på fem lande koordineret af Germán J. de Valcárcel Gonzalvo, professor i optik ved universitetet i Valencia, har udviklet en -
 Test af Einsteins ækvivalensprincip nær et supermassivt sort hulBillede af Galactic Center. Kredit:European Southern Observatory (ESO). GRAVITY -samarbejdet, et team af forskere på flere kendte institutter, herunder Max Planck Institute, LESIA Paris Observator
Test af Einsteins ækvivalensprincip nær et supermassivt sort hulBillede af Galactic Center. Kredit:European Southern Observatory (ESO). GRAVITY -samarbejdet, et team af forskere på flere kendte institutter, herunder Max Planck Institute, LESIA Paris Observator -
 Æggekarton quantum dot array kan føre til ultralav strømforsyningsenhederKredit:Unsplash/CC0 Public Domain En ny vej mod at sende og modtage information med enkeltfotoner af lys er blevet opdaget af et internationalt team af forskere under ledelse af University of Mich
Æggekarton quantum dot array kan føre til ultralav strømforsyningsenhederKredit:Unsplash/CC0 Public Domain En ny vej mod at sende og modtage information med enkeltfotoner af lys er blevet opdaget af et internationalt team af forskere under ledelse af University of Mich -
 Undersøgelse afslører nye detaljer om, hvad der skete i det første mikrosekund af Big BangKredit:CC0 Public Domain Forskere fra Københavns Universitet har undersøgt, hvad der skete med en bestemt slags plasma - det første stof, der nogensinde har været til stede - i løbet af det første
Undersøgelse afslører nye detaljer om, hvad der skete i det første mikrosekund af Big BangKredit:CC0 Public Domain Forskere fra Københavns Universitet har undersøgt, hvad der skete med en bestemt slags plasma - det første stof, der nogensinde har været til stede - i løbet af det første
- En vulkanologs udsigt over Kilauea
- Forskere skaber kunstige materialer atom-for-atom
- Sporing af eksploderende isrevner på Himalaya-gletsjere
- Hvad blev Silt brugt til i det gamle Egypten?
- Ny forskning viser, at grafen er i stand til automatisk at lukke huller i sig selv
- Et 3D-printet teleskop:Den analoge skydrifter