


Hvordan computere søger efter fremtidens stoffer

Kredit:University of California, San Francisco
Opdagelse af lægemidler kan bringe tankerne hen på billeder af hvide laboratoriefrakker og pipetter, men da Henry Lin, PhD, for nylig satte sig for at finde et bedre opioid med færre bivirkninger, hans første skridt var at tænde for computere.
Ved at bruge et program kaldet DOCK, han uploadede en krystalstruktur af opioidreceptoren fundet i hjernen og fik adgang til et virtuelt bibliotek med 3 millioner forbindelser, der kunne binde sig til en kemisk "lomme" på receptoren. De fleste lægemidler – fra antibiotika til antidepressiva – virker ved at binde sig til specifikke steder på proteiner, men for at være effektiv, de skal passe helt rigtigt.
Programmet drejede hver forbindelse rundt, overvejede fleksibiliteten af dets forskellige vedhæng, og efter at have testet et gennemsnit på 1,3 millioner konfigurationer pr. forbindelse – rangeret dem efter deres bindingspotentiale. Processen, kører på computere, der er tilsluttet kraftige processorer, tog omkring to uger.
En kandidatstuderende på det tidspunkt, Lin arbejdede sammen med sin rådgiver Brian Shoichet, PhD, professor i farmaceutisk kemi ved UC San Francisco School of Pharmacy, og Aashish Manglik, PhD, fra Stanford University for at finde top 2, 500 forbindelser for yderligere faktorer og udvalgt 23 til eksperimentel testning i levende celler - cue laboratoriefrakker og pipetter.
I stigende grad, forskere henvender sig til virtuelle eksperimenter for de indledende trin af lægemiddeludvikling. Med stadig hurtigere computere, den tidlige og overvejende trial-and-error fase af lægemiddeludvikling kan reduceres til et spørgsmål om dage, og med stadigt voksende onlinebiblioteker af forbindelser, lægemiddelskærme kan omfatte, bogstaveligt talt, al den kendte kemi i verden.
Styrker og begrænsninger
Forskere er forsigtige med hensyn til computeropdagelsens potentiale - kun en lille del af lovende forbindelser virker faktisk, når de testes i det virkelige liv - men de siger, at en af dens styrke er at afsløre helt nye forbindelser som lægemiddelkandidater.
Shoichet har specialiseret sig i en populær beregningsmetode kendt som molekylær docking. "Hvor docking passer ind er i tidlig opdagelsesforskning, i at finde nye afgange, " han sagde.
Hans teams søgen efter det nye opioid illustrerer både styrkerne og begrænsningerne ved opdagelse af computermedicin.
Faktisk, de oprindelige opioidkandidater identificeret gennem molekylær docking udførte kun beskedent i eksperimentel test. "Stadig, den aktivitet, de havde, var meget reproducerbar, og molekylerne var meget nye, varsler om ny biologi, " sagde Shoichet.
Holdet dockede endnu en runde af forbindelser med lignende strukturer og testede topscorerne. Med samarbejdspartnere ved University of North Carolina, Chapel Hill og Friedrich Alexander Universitet i Tyskland, de identificerede den mest potente forbindelse og optimerede dens farmakologi med computerstyret syntetisk udarbejdelse.
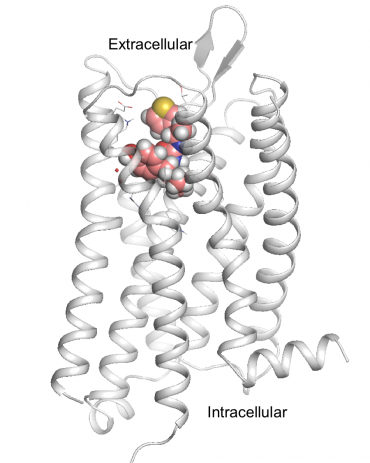
PZM21, den nye, sikrere opioidlægemiddelkandidat, er vist forankret på hjernens morfinreceptor, mu-opioid-receptoren. Kredit:Anat Levit
Den vindende forbindelse, navngivet PZM21, er kemisk ulig nogen i nuværende brug og er muligvis ikke blevet fundet gennem mere traditionelle metoder. Det er en fuldstændig beregningsmæssigt designet forbindelse, der er mere potent end morfin. Hos mus, det blokerede effektivt smerte uden de sædvanlige bivirkninger af respirationsundertrykkelse og forstoppelse og så endda ud til at være mindre vanedannende.
Docking er ikke en sølvkugle, men det er blevet et stærkt startpunkt i lang tid, tværfaglig proces med lægemiddeludvikling. Blandt dets vigtigste bidrag har været proteasehæmmere, der har hjulpet med at gøre HIV til en sygdom, der kan behandles. Forskere bruger også docking til at screene lægemiddelkandidater til behandling af brystkræft, hepatitis C, forhøjet blodtryk, stafylokokker, SARS-virus og influenza.
Teknologi banebrydende på UCSF
Molekylær docking blev pioneret for tre årtier siden af en ung UCSF fysisk kemiker ved navn Tack Kuntz, PhD, nu professor emeritus ved Farmaciskolen. Da Kuntz ankom til campus i begyndelsen af 1970'erne, den traditionelle tilgang til lægemiddelopdagelse sejrede stadig.
Som Kuntz beskrev det, processen var afhængig af tilfældigheder og meget lidt teori:"Du går ud og finder nye naturlige forbindelser og bringer dem tilbage for at teste i et laboratorium. Bare sæt kemikalier sammen med en organisme og se, hvad der sker."
Farmaceutiske kemikere skænkede næppe en tanke til de molekylære detaljer om, hvordan lægemidler interagerer med kroppen. Mange stoffer, inklusive de første antibiotika, var blevet opdaget serendipitalt, men Kuntz, efter at have set den nye molekylære forståelse skylle over biologiens felt, følte, at det var tid til en lignende opdatering inden for farmakologi.
"Det målbaserede syn på biologi - at man kan forstå biologi gennem uafhængige proteiner og genprodukter - havde allerede overtaget, men farmakologi var et årti bagud, " sagde Shoichet, som var kandidatstuderende i Kuntz' laboratorium i 1980'erne.
Kuntz og hans kolleger begyndte at arbejde hen imod en mere rationel tilgang til lægemiddeldesign, hvor de forsøgte at identificere forbindelser, der kunne passe til specifikke receptorer på proteiner, som at finde den manglende brik i et puslespil. I 1982, de udgav et papir, der beskrev det første molekylære docking-program, der kunne "udforske geometrisk mulige justeringer af ligander og receptorer med kendt struktur."
Kuntz sendte 10, 000 eksemplarer af det første docking-program til forskere rundt om i landet. Snart, andre forskere var ved at udvikle lignende beregningsprogrammer, og begejstringen spredte sig hurtigt uden for den akademiske verden. I 1990'erne, alle større medicinalvirksomheder havde åbnet en enhed til computeropdagelse af lægemidler.
Indhente en idé
På trods af den indledende begejstring, imidlertid, Opdagelse af computermedicin førte ikke til hurtige resultater. Kuntz' idé var kommet forud for sin tid. Det ville tage årtier med trinvise fremskridt inden for molekylærbiologi, billed- og computerteknologi, før opdagelse af computermedicin kunne begynde at opfylde sit løfte.

Tack Kuntz, PhD, og hans kolleger i 1982 udgav et papir, der beskrev det første molekylære docking-program, der kunne "udforske geometrisk mulige justeringer af ligander og receptorer med kendt struktur." Kredit:University of California, San Francisco
En væsentlig begrænsning i 1990'erne var manglen på kendte proteinstrukturer. Uden disse, der var få mål at finde stoffer til. I årtierne siden, tusindvis af proteinstrukturer af mulige lægemiddelmål er blevet afsløret ved røntgenkrystallografi og nuklear magnetisk resonansbilleddannelse.
Opdagelsen af den nye opioidkandidat, for eksempel, var kun muligt på grund af de nyligt bestemte krystalstrukturer af G-protein-koblede receptorer, en familie af proteiner, der inkluderer opioidreceptoren.
Virtuelle biblioteker af forbindelser er også vokset eksponentielt. I 1991, en database kan indeholde 55, 000 forbindelser; nu indeholder de titusindvis af millioner. "Omfanget af den kemi, vi prøver, er steget omkring samme hastighed som Moores lov, " sagde Shoichet. "Der er en umættelig hunger efter flere og flere molekyler."
Dagens docking-programmer er i stand til realistisk at modellere interaktionerne på atomniveau mellem et lægemiddel og dets mål, men nogle vanskelige detaljer – såsom hvordan atomkræfter ændrer sig, når et lægemiddelmolekyle fortrænger vand på bindingsstedet – er fortsat igangværende udfordringer på området.
Løfter og beviser
Molekylær docking er ikke den eneste form for computerbaseret lægemiddeldesign. På UCSF Institute for Computational Health Sciences (ICHS), snesevis af forskere udforsker utallige beregningsmetoder til at fremme medicinsk forskning.
Michael Keiser, PhD, et medlem af ICHS og en adjunkt ved Institute of Neurodegenerative Diseases, studerer lægemidler, der rammer mange molekylære mål på én gang, som om at slå en akkord i stedet for en enkelt tone. Denne multi-target handling var længe forstået at være årsagen til utilsigtede bivirkninger, men kan også rettes mod at behandle komplekse sygdomme.
Først i begyndelsen af 2000'erne kom forskerne til at erkende, at mange eksisterende lægemidler virker gennem mere end ét mål - antipsykotika, for eksempel, der rammer både serotonin- og dopaminreceptorer. De designer nu med vilje stoffer til at gøre det.
"For nogle sygdomme, der endnu ikke har behandling, måske er det fordi der ikke er et eneste protein, du skal tænde eller slukke for; hvad hvis stoffet skal ramme flere mål i stedet?" sagde Keiser, som var kandidatstuderende hos Shoichet.
I sit laboratorium, Keizer bruger beregningsmetoder til at identificere kemiske mønstre blandt lægemidler, der binder til det samme sæt af mål og finde nye forbindelser, der har matchende farmakologi. Denne beregningsmetode kan genkende ligheder mellem forbindelser, som mere konventionelle analyser ville gå glip af. Keizer søger nu mod kunstig intelligens-teknologi, kendt som deep learning, for endnu bedre mønstergenkendelse.
Selv når beregningsmetoder tager fart, deres bevis er stadig i den virkelige verden - i celler, dyremodeller, og i sidste ende i klinikken. "I et stykke tid var det almindeligt at udgive artikler med forudsigelser om et lille molekyles aktiviteter, men ingen egentlig test af disse forudsigelser, fordi eksperimenterne for at gøre det var dyre, vanskelig eller esoterisk, sagde Keiser.
Efterhånden som behovet for samarbejde er blevet tydeligt, partnerskabet mellem beregningsforudsigelse og våde laboratorieeksperimenter er blevet mærkbart styrket i det sidste årti, sagde Keiser. "Trods alt, hvordan kan du forbedre dine forudsigelser, hvis du ikke er sikker på, hvilke der er forkerte?"
 Varme artikler
Varme artikler
-
 Forskere designer sensorer til hurtigt at opdage plantehormonerNanosensorer udviklet ved Singapore-MIT Alliance for Research and Technology (SMART) kan detektere syntetiske auxin plantehormoner NAA og 2, 4-D. Kredit:Singapore-MIT Alliance for Research and Technol
Forskere designer sensorer til hurtigt at opdage plantehormonerNanosensorer udviklet ved Singapore-MIT Alliance for Research and Technology (SMART) kan detektere syntetiske auxin plantehormoner NAA og 2, 4-D. Kredit:Singapore-MIT Alliance for Research and Technol -
 Lys til litografi kunne passere trykte fibreFig. UV-elektromagnetiske bølger, der passerer gennem den trykte fiber, kan nå målfotoresisten. Kredit:University of Utah University of Utah forskere har udviklet et trykt fiberbaseret lysmoduleri
Lys til litografi kunne passere trykte fibreFig. UV-elektromagnetiske bølger, der passerer gennem den trykte fiber, kan nå målfotoresisten. Kredit:University of Utah University of Utah forskere har udviklet et trykt fiberbaseret lysmoduleri -
 Forskere udvikler fuldt soldrevet autonomt kemisk minianlægDen solcelledrevne minireaktor. Kredit:Noël Research Group Professor Timothy Noël og kolleger i Flow Chemistry-gruppen ved Van t Hoff Instituttet for Molekylær Videnskab ved Universitetet i Amster
Forskere udvikler fuldt soldrevet autonomt kemisk minianlægDen solcelledrevne minireaktor. Kredit:Noël Research Group Professor Timothy Noël og kolleger i Flow Chemistry-gruppen ved Van t Hoff Instituttet for Molekylær Videnskab ved Universitetet i Amster -
 Nye ringslutningsreaktioner til syntetisering af makrocykliske lægemiddelledningerIntegreret robotarbejdsstation brugt på EPFLs Biomolecular Screening Facility til at udføre kombinatorisk syntese af makrocykler og efterfølgende high-throughput målbaserede screeningsassays. Kredit:A
Nye ringslutningsreaktioner til syntetisering af makrocykliske lægemiddelledningerIntegreret robotarbejdsstation brugt på EPFLs Biomolecular Screening Facility til at udføre kombinatorisk syntese af makrocykler og efterfølgende high-throughput målbaserede screeningsassays. Kredit:A
- Design af dagens byer til morgendagens udfordringer
- Hvorfor bliver dåse luft kold?
- Global opvarmning, El Nino kan forårsage vådere vintre, tørrere forhold i andre måneder
- Erfaringer fra Sierra Leones Ebola-pandemi om virkningen af skolelukninger på piger
- Satellitkort registrerer oversvømmelser i Australien
- Effektivitetsspring ved adskillelse af para-xylen ved hjælp af nye kulstofmembraner