


Energiniveaujustering for molekylær elektronik
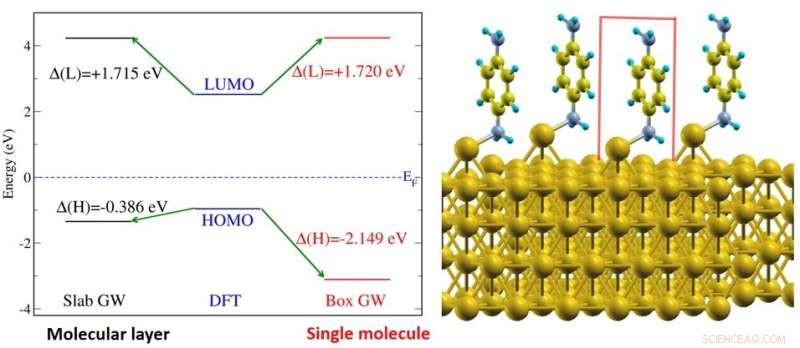
(Venstre) Figur viser elektronenerginiveaujusteringen af benzen-diaminmolekyler på guldoverfladesystemet (vist til højre). Energiniveauerne er vist for et molekylært lag (sort) og for et enkelt molekyle (rødt). (Til højre) Illustration af benzen-diamin-molekylerne på guldoverfladen. Kredit:National University of Singapore
NUS-fysikere har fundet ud af, at komplekse elektron-elektron-interaktioner ændrer energiniveauerne ved molekyle-metal-grænseflader, påvirker ydeevnen af molekylært elektronisk udstyr.
Molekylær elektronik involverer brugen af molekyler som den vigtigste byggesten til at skabe det elektroniske kredsløb. Det kan potentielt bruges til at udvikle kredsløb, der er meget mindre end dem, der er lavet af konventionelle siliciumprocesser. Forståelse af de elektroniske egenskaber af grænsefladen mellem molekylerne og metallederne, især deres tilknyttede energiniveauer, er vigtig for at rationalisere og optimere enhedens ydeevne. Dette er centralt for udviklingen af molekylær elektronik.
En grundlæggende egenskab ved hvert molekyle er dets energigab, defineret som energiforskellen mellem det højeste og laveste orbitale energiniveau optaget og ubesat af henholdsvis elektroner. Disse niveauer er også de vigtigste energiniveauer for enhedens ydeevne. Energigabet i et molekyle bliver mindre, når molekylet bringes tæt på en metaloverflade; dette vil gøre det lettere for ladningsbærere at bevæge sig mellem molekylet og metalkontakten. Denne ændring i mellemrummet er primært forårsaget af elektroniske screeningseffekter fra metaloverfladen, og kan være så stor som adskillige elektron-volt. Imidlertid, denne elektroniske screeningseffekt mangler i de fleste teoretiske undersøgelser om dette emne.
Et forskerhold ledet af prof Su Ying QUEK, fra Institut for Fysik, NUS har belyst grænsefladens elektroniske strukturegenskaber for en række forskellige molekyler på guldoverflader ved hjælp af state-of-the-art teoretiske og beregningsmetoder, der eksplicit tager højde for elektroniske screeningseffekter fra de første principper. Forskerne udførte beregninger på molekylære systemer forankret af almindelige kemiske funktionelle grupper (amin, pyridin- og thiolatgrupper). Forskerholdet fandt, at for et enkelt molekyle, den elektroniske screeningseffekt kan forudsiges nøjagtigt ud fra en billedladningsmodel, selv i nærværelse af kemiske bindinger. Billedladningsmodellen er en klassisk elektrostatisk metode, som tilnærmer den elektroniske screening af en testladning med en billedladning i metallet. Imidlertid, i enheder med mange molekyler, forskerne fandt betydelige yderligere elektroniske screeningsmekanismer. Udover intermolekylære screeningseffekter, substrat-medierede intermolekylære interaktioner har også vist sig at bidrage til disse yderligere screeningsmekanismer. Resultaterne tyder på, at ladningsbærere lettere kan tunnelere over grænsefladen i enheder med mange molekyler.
Prof Quek sagde, "Dette arbejde giver værdifuld indsigt i de mange elektroneffekter ved molekyle-metal-grænsefladerne, der involverer kemiske bindinger. Resultaterne og resultaterne fra denne forskning udgør et vigtigt skridt i retning af forståelse og manipulation af funktionelle organiske systemer i udviklingen af molekylære enheder."
 Varme artikler
Varme artikler
-
 Under pres:Nyt bioinspireret materiale kan formskifte til eksterne kræfterTil JHU-teamets eksperiment, øget kraft (pilen peger nedad) påført materialet førte til flere elektriske ladninger, og dermed, mere mineralisering. Kredit:Pam Li/Johns Hopkins University Inspirere
Under pres:Nyt bioinspireret materiale kan formskifte til eksterne kræfterTil JHU-teamets eksperiment, øget kraft (pilen peger nedad) påført materialet førte til flere elektriske ladninger, og dermed, mere mineralisering. Kredit:Pam Li/Johns Hopkins University Inspirere -
 Vedvarende energi kan reducere udstødningsemissioner drastiskEn kompleks opsætning af objektiver og kameraer er påkrævet for direkte, laserbaserede NOx-målinger i en flamme. Kredit:© 2018 Miles Bohon Skift til vedvarende brændstoffer kan reducere udstødning
Vedvarende energi kan reducere udstødningsemissioner drastiskEn kompleks opsætning af objektiver og kameraer er påkrævet for direkte, laserbaserede NOx-målinger i en flamme. Kredit:© 2018 Miles Bohon Skift til vedvarende brændstoffer kan reducere udstødning -
 Hvad sker der, når du tilføjer et fald madfarve til koldt vand?Fødevarer farvning illustrerer diffusion i vand. Diffusion er blanding af molekyler på grund af deres tilfældige bevægelse, uanset om de er i en væske eller en gas. Fordi molekyler i koldt vand har mi
Hvad sker der, når du tilføjer et fald madfarve til koldt vand?Fødevarer farvning illustrerer diffusion i vand. Diffusion er blanding af molekyler på grund af deres tilfældige bevægelse, uanset om de er i en væske eller en gas. Fordi molekyler i koldt vand har mi -
 Min ambition? En anden nobelpris siger kemipristagerCharpentier modtog sit Nobeldiplom og medalje af Per Thoresson, den svenske ambassadør i Tyskland, i Berlin på mandag At vinde Nobelprisen er ofte toppen af professionel præstation, men kemipris
Min ambition? En anden nobelpris siger kemipristagerCharpentier modtog sit Nobeldiplom og medalje af Per Thoresson, den svenske ambassadør i Tyskland, i Berlin på mandag At vinde Nobelprisen er ofte toppen af professionel præstation, men kemipris
- Fysikere måler mekaniske egenskaber af 2-D monolag materialer
- Hvordan man laver en 3D-model af et atom
- Observatorium på størrelse med en galakse ser potentielle antydninger af gravitationsbølger
- Globale beviser forbinder stigning i ekstrem nedbør til menneskedrevne klimaændringer
- En ny kunstig neurale netværksramme for gangbaseret biometri
- Filippinerne lukker Boracay for turister under høj sikkerhed