


Automatisering af molekyledesign for at fremskynde lægemiddeludvikling
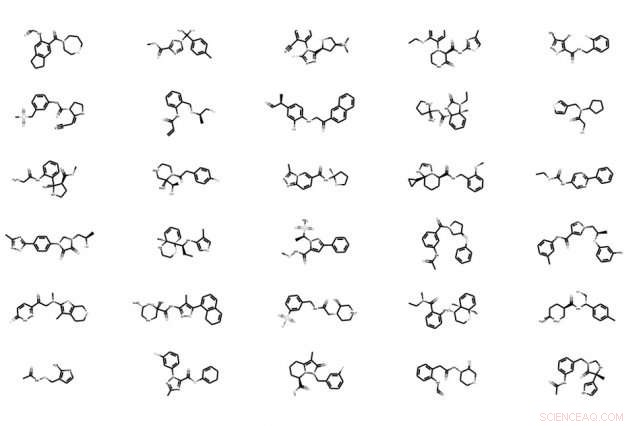
MIT-forskere har udviklet en maskinlæringsmodel, der bedre udvælger molekylekandidater til terapi, samtidig med at det giver mulighed for automatiseret modifikation af den molekylære struktur for højere styrke. Innovationen har potentiale til at fremskynde udviklingen af lægemidler. Kredit:Massachusetts Institute of Technology
Design af nye molekyler til lægemidler er primært en manual, tidskrævende proces, der er tilbøjelig til at fejle. Men MIT-forskere har nu taget et skridt i retning af at fuldautomatisere designprocessen, hvilket kunne fremskynde tingene drastisk - og give bedre resultater.
Lægemiddelopdagelse er afhængig af blyoptimering. I denne proces, kemikere vælger et mål ("bly") molekyle med kendt potentiale til at bekæmpe en specifik sygdom, tweak derefter dens kemiske egenskaber for højere styrke og andre faktorer.
Tit, kemikere bruger ekspertviden og udfører manuel justering af molekyler, tilføje og trække funktionelle grupper - atomer og bindinger, der er ansvarlige for specifikke kemiske reaktioner - en efter en. Selvom de bruger systemer, der forudsiger optimale kemiske egenskaber, kemikere skal stadig udføre hvert modifikationstrin selv. Dette kan tage timer for hver iteration og producerer muligvis stadig ikke en gyldig lægemiddelkandidat.
Forskere fra MIT's Computer Science and Artificial Intelligence Laboratory (CSAIL) og Department of Electrical Engineering and Computer Science (EECS) har udviklet en model, der bedre udvælger blymolekylekandidater baseret på ønskede egenskaber. Det ændrer også den molekylære struktur, der er nødvendig for at opnå en højere styrke, samtidig med at det sikres, at molekylet stadig er kemisk gyldigt.
Modellen tager dybest set som input molekylære strukturdata og skaber direkte molekylære grafer - detaljerede repræsentationer af en molekylær struktur, med noder, der repræsenterer atomer, og kanter, der repræsenterer bindinger. Det opdeler disse grafer i mindre klynger af gyldige funktionelle grupper, som det bruger som "byggesten", der hjælper det mere nøjagtigt at rekonstruere og bedre modificere molekyler.
"Motivationen bag dette var at erstatte den ineffektive menneskelige modifikationsproces med at designe molekyler med automatiseret iteration og sikre gyldigheden af de molekyler, vi genererer, " siger Wengong Jin, en ph.d. studerende i CSAIL og hovedforfatter på et papir, der beskriver modellen, der præsenteres på 2018 International Conference on Machine Learning i juli.
Med Jin på papiret er Regina Barzilay, Delta Electronics Professor ved CSAIL og EECS og Tommi S. Jaakkola, Thomas Siebel professor i elektroteknik og datalogi i CSAIL, EECS, og på Institut for Data, Systemer, og samfundet.
Forskningen blev udført som en del af Machine Learning for Pharmaceutical Discovery and Synthesis Consortium mellem MIT og otte farmaceutiske virksomheder, annonceret i maj. Konsortiet identificerede leadoptimering som en nøgleudfordring i lægemiddelopdagelse.
"I dag, det er virkelig et håndværk, hvilket kræver en masse dygtige kemikere for at få succes, og det er det, vi ønsker at forbedre, " siger Barzilay. "Det næste skridt er at tage denne teknologi fra den akademiske verden til brug på rigtige farmaceutiske designsager, og demonstrere, at det kan hjælpe menneskelige kemikere med at udføre deres arbejde, hvilket kan være udfordrende."
"At automatisere processen giver også nye maskinlæringsudfordringer, " siger Jaakkola. "At lære at relatere, modificere, og generere molekylære grafer driver nye tekniske ideer og metoder."
Generering af molekylære grafer
Systemer, der forsøger at automatisere molekyledesign, er dukket op i de seneste år, men deres problem er gyldighed. Disse systemer, Jin siger, genererer ofte molekyler, der er ugyldige under kemiske regler, og de formår ikke at producere molekyler med optimale egenskaber. Dette gør i det væsentlige fuld automatisering af molekyledesign umulig.
Disse systemer kører på lineære notationer af molekyler, kaldet "forenklede molekylære input line-entry-systemer, " eller SMIL, hvor lange rækker af bogstaver, tal, og symboler repræsenterer individuelle atomer eller bindinger, der kan fortolkes af computersoftware. Når systemet modificerer et blymolekyle, den udvider sin strengrepræsentation symbol for symbol - atom for atom, og binding for binding – indtil den genererer en endelig SMILES-streng med højere styrke af en ønsket egenskab. Til sidst, systemet kan producere en endelig SMILES-streng, der virker gyldig under SMILES-grammatik, men er faktisk ugyldig.
Forskerne løser dette problem ved at bygge en model, der kører direkte på molekylære grafer, i stedet for SMILES-strenge, som kan ændres mere effektivt og præcist.
Modellen drives af en tilpasset variationsautoencoder - et neuralt netværk, der "koder" et inputmolekyle ind i en vektor, som dybest set er et lagerplads for molekylets strukturelle data, og derefter "afkoder" den vektor til en graf, der matcher input-molekylet.
I indkodningsfasen, modellen opdeler hver molekylær graf i klynger, eller "undergrafer, " som hver repræsenterer en specifik byggeklods. Sådanne klynger er automatisk konstrueret af et fælles maskinlæringskoncept, kaldet trænedbrydning, hvor en kompleks graf er kortlagt i en træstruktur af klynger - "hvilket giver et stillads af den originale graf, " siger Jin.
Både stilladstræstruktur og molekylær grafstruktur er kodet ind i deres egne vektorer, hvor molekyler er grupperet efter lighed. Dette gør det lettere at finde og modificere molekyler.
I afkodningsfasen, modellen rekonstruerer den molekylære graf på en "grov-til-fin" måde - gradvist stigende opløsning af et lavopløsningsbillede for at skabe en mere raffineret version. Det genererer først det træstrukturerede stillads, og samler derefter de tilhørende klynger (knuder i træet) sammen til en sammenhængende molekylær graf. Dette sikrer, at den rekonstruerede molekylære graf er en nøjagtig replikation af den oprindelige struktur.
For leadoptimering, modellen kan derefter modificere blymolekyler baseret på en ønsket egenskab. Det gør det ved hjælp af en forudsigelsesalgoritme, der scorer hvert molekyle med en styrkeværdi for den egenskab. I avisen, for eksempel, forskerne søgte molekyler med en kombination af to egenskaber - høj opløselighed og syntetisk tilgængelighed.
Givet en ønsket ejendom, modellen optimerer et hovedmolekyle ved at bruge forudsigelsesalgoritmen til at modificere dets vektor – og, derfor, struktur - ved at redigere molekylets funktionelle grupper for at opnå en højere styrkescore. Det gentager dette trin i flere gentagelser, indtil den finder den højeste forudsagte styrkescore. Derefter, modellen afkoder endelig et nyt molekyle fra den opdaterede vektor, med ændret struktur, ved at kompilere alle de tilsvarende klynger.
Gyldig og mere potent
Forskerne trænede deres model på 250, 000 molekylære grafer fra ZINC-databasen, en samling af 3-D molekylære strukturer tilgængelige til offentlig brug. De testede modellen på opgaver til at generere gyldige molekyler, finde de bedste blymolekyler, og designe nye molekyler med øget styrke.
I den første test, forskernes model genererede 100 procent kemisk gyldige molekyler fra en prøvefordeling, sammenlignet med SMILES-modeller, der genererede 43 procent gyldige molekyler fra samme fordeling.
Den anden test involverede to opgaver. Først, modellen gennemsøgte hele samlingen af molekyler for at finde det bedste blymolekyle for de ønskede egenskaber - opløselighed og syntetisk tilgængelighed. I den opgave modellen fandt et blymolekyle med en 30 procent højere styrke end traditionelle systemer. Den anden opgave involverede at modificere 800 molekyler for højere styrke, men er strukturelt ligner blymolekylet. Derved, modellen skabte nye molekyler, ligner meget ledningens struktur, i gennemsnit mere end 80 procent forbedring i styrke.
Forskerne sigter derefter mod at teste modellen på flere egenskaber, ud over opløselighed, som er mere terapeutisk relevante. At, imidlertid, kræver flere data. "Farmaceutiske virksomheder er mere interesserede i ejendomme, der kæmper mod biologiske mål, men de har færre data om dem. En udfordring er at udvikle en model, der kan arbejde med en begrænset mængde træningsdata, " siger Jin.
Denne historie er genudgivet med tilladelse fra MIT News (web.mit.edu/newsoffice/), et populært websted, der dækker nyheder om MIT-forskning, innovation og undervisning.
Sidste artikelForskere bruger fotoner til at adskille metalioner
Næste artikelNy verdensrekord for direkte solvandsspaltningseffektivitet
 Varme artikler
Varme artikler
-
 Metal hvisken:At finde en bedre måde at genvinde ædle metaller fra elektronisk affaldNy teknologi udviklet af Iowa State ingeniører bruger varme og oxidation til at genvinde rene og ædle metaller fra elektronisk affald. Det virker på to måder - det kan bringe de mest reaktive komponen
Metal hvisken:At finde en bedre måde at genvinde ædle metaller fra elektronisk affaldNy teknologi udviklet af Iowa State ingeniører bruger varme og oxidation til at genvinde rene og ædle metaller fra elektronisk affald. Det virker på to måder - det kan bringe de mest reaktive komponen -
 Ny tilgang forudsiger glas altid udviklende adfærd ved forskellige temperaturerMultiskala modellering af et polymerglas for at forudsige dets temperaturafhængige egenskaber. Kredit:Wenjie Xia/NIST Ikke alt ved glas er klart. Hvordan dets atomer er arrangeret og opfører sig,
Ny tilgang forudsiger glas altid udviklende adfærd ved forskellige temperaturerMultiskala modellering af et polymerglas for at forudsige dets temperaturafhængige egenskaber. Kredit:Wenjie Xia/NIST Ikke alt ved glas er klart. Hvordan dets atomer er arrangeret og opfører sig, -
 Sådan bestemmes den højeste ioniseringsenergiNår det kommer til kemi, er det svært at forestille sig et mere kendt billede end en tæt pakket kerne af protoner og neutroner omgivet af elektroner i deres orbitaler. Hvis du har brug for at sammenli
Sådan bestemmes den højeste ioniseringsenergiNår det kommer til kemi, er det svært at forestille sig et mere kendt billede end en tæt pakket kerne af protoner og neutroner omgivet af elektroner i deres orbitaler. Hvis du har brug for at sammenli -
 Hvordan bittesmå enzymer troner øverst i verdensomspændende kulstofgenbrugHvide rådsvampe, Duke Forest, North Carolina Kredit:NA Genanvendelsen af det meste af kulstoffet i naturen afhænger af nedbrydning af to polymerer i træemateriale, især cellulose og lignin. I et
Hvordan bittesmå enzymer troner øverst i verdensomspændende kulstofgenbrugHvide rådsvampe, Duke Forest, North Carolina Kredit:NA Genanvendelsen af det meste af kulstoffet i naturen afhænger af nedbrydning af to polymerer i træemateriale, især cellulose og lignin. I et
- Sextuply-formørkende sekstuppelstjernesystem afdækket i TESS-data med en assist fra AI
- Nano-guitarstrengen, der spiller sig selv
- Miljøpåvirkning af flaskevand op til 3, 500 gange højere end ledningsvand
- Fang energi fra sollys med farvestoffer inspireret af naturen
- Dødstal fra oversvømmelser i Sydasien topper 1, 000
- Nevada overvejer teknologi til at scanne mobiltelefoner efter nedbrud