


Forskere finder en ny måde at målrette mod influenzavirus

Rice University og Baylor College of Medicine forskere brugte computersimuleringer til at studere den proces, hvorved hæmagglutinin hjælper vira med at invadere og inficere celler. Forskerne mener, at proteinets stamdomæne udfolder sig og foldes igen til en anden konfiguration, når det udløses, men holder pause for at frigive et skjult fusionspeptid, der binder virussen til målcellen. Klik på billedet for en større version. Kredit:Xingcheng Lin
Der er et problem i svinget med et protein, der afgiver influenzavirus. Forskere fra Rice University og Baylor College of Medicine mener, at denne mekanisme kan være et nyttigt mål for at forhindre virussen i at inficere celler.
I et papir i Proceedings of the National Academy of Sciences, Rice-Baylor-teamet ledet af biofysiker José Onuchic og biokemikerne Jianpeng Ma og Qinghua Wang dykker yderligere ned i et glycoproteinkompleks, som det begyndte at definere i et papir fra 2014.
Det protein, hæmagglutinin, sidder på overfladen af influenzavirus og hjælper dem med at binde sig til og transporteres gennem målcellernes beskyttende membraner.
Papiret begynder at definere mekanismen, der gør det muligt for proteinet at folde sig ud og foldes igen på et øjeblik, ændre sin form for at afsløre et peptid, der binder virussen til en celle og begynder infektion. Forskerne mener, at terapeutiske lægemidler kan bruge denne mekanisme til at lukke virussen ned.
"Dette protein starter i en foldet tilstand og gennemgår en global transformation, foldes igen i en helt anden tilstand, " sagde Onuchic, meddirektør for Rice's Center for Teoretisk Biologisk Fysik (CTBP). "Men der er en lille del i centrum, som evolutionen har bevaret."
Den enkelte bevarede aminosyrerest er det problem, der får proteinet til at holde pause i genfoldningsprocessen. Det tillader et fusionspeptid, der er begravet indeni, at binde sig til målcellen og begynde at inficere den. Uden pause, genfoldningen ville være for hurtig til, at bindingen kunne finde sted.
Hovedforfatter og Rice postdoc-forsker Xingcheng Lin modellerede den del af proteinet, B-løkken i HA2-domænet. HA2 sidder under et andet domæne, en kasket kendt som HA1, der muterer for at undslippe tidligere forsvar. Lin forklarede, at HA1 er et almindeligt mål for influenzamedicin, fordi det eksponerede cap-domæne er mere tilgængeligt end det beskyttede HA2-domæne.
Problemet er, at HA1 konstant muterer for at modstå medicin, han sagde. Det påvirker, hvor effektive influenzavacciner er hvert år. Lin og Onuchic sagde, at HA2 præsenterer et bedre mål for lægemidler, fordi mekanismen er meget konserveret af evolution.
"Hvis et lægemiddel er rettet mod HA2, domænet kan ikke undslippe ved at lave mutationer, fordi mutationerne i sig selv ville gøre det ikke-funktionelt, " sagde Lin. "Den slags medicin kunne blive en universel vaccine."
HA2 er en trimer struktur, der, når det udløses af sure forhold i miljøet nær en målcelle, transformerer sig selv fra en tilfældig sløjfe til en oprullet spole. Selv med pausen, den folder sig ud og foldes igen på en brøkdel af et sekund, alt for hurtigt til at mikroskoper kan se. Men en computersimulering af processen kan bremses.
Det er tilfældigvis en specialitet for CTBP, som bruger programmer, der analyserer proteiners energilandskab til at forudsige, hvordan de vil foldes. Onuchic og hans kolleger er pionerer inden for teorien om, at foldeproteiner følger en ordnet, "kanaliseret" proces, der afhænger af den iboende energi af hvert atom i kæden, som hver konstant søger sin laveste energitilstand. Hvis alle atomare "perler" kan identificeres, det er muligt at simulere den komplekse foldeproces.
Risforskerne bruger ofte grovkornede modeller af proteiner, en delmængde af atomer, der repræsenterer helheden, at forudsige, hvordan de vil folde. Den nye undersøgelse var meget mere ambitiøs og havde til formål at forudsige den komplekse udfoldelse og genfoldning ved at bruge ikke kun hvert atom i kæden, men også hvert atom i dets flydende miljø, sagde Onuchic.
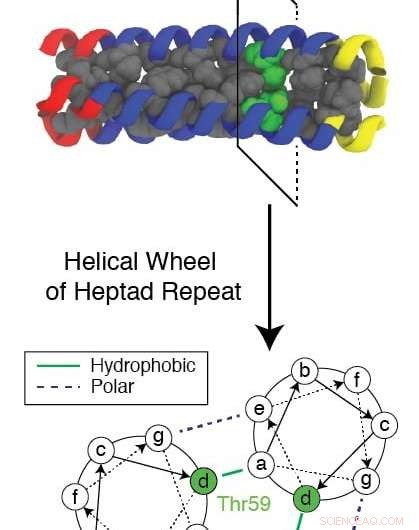
En evolutionært konserveret rest kendt som Thr59 forstyrrer det gentagne mønster, der dannes af et trimerisk protein, når det foldes igen, mens det hjælper en influenzavirus med at inficere en celle. Forskere ved Rice University og Baylor College of Medicine brugte en kompleks computersimulering til at studere mekanismen og lede efter nye mål for lægemidler til at stoppe influenza. Kredit:Xingcheng Lin
Lin modellerede 40 mikrosekunder (milliontedele af et sekund) af HA2-domæneovergangen, der repræsenterer hele processen, som tager 1,4 millisekunder (tusindedele af et sekund) at fuldføre. Selv den forkortede proces tog to års computertid at levere resultater, han sagde.
"Det simulerede domæne er omkring 3, 000 atomer, men når miljøet, inklusive vand, er redegjort for, den samlede simulering omfatter omkring 100, 000 atomer, " sagde Onuchic. "Det er stadig en enorm simulering, der krævede state-of-the-art teknikker."
Tidligere teorier baseret på krystallografiske billeder af før-og-efter-proteinerne fremlagde ideen om et fjederbelastet domæne, der så ud til at hæfte sig til målcellen efter hættens fjernelse. Onuchic sagde, at den komplette model af HA2 understøtter en anden mekanisme.
"Vi fandt ud af, at der er en masse energi, der gør den endelige tilstand af HA2 meget mere stabil end den oprindelige tilstand, " sagde han. "Men med den fjederbelastede mekanisme, det meste af energien ville allerede være spildt, når den danner den oprullede spiral og binder cellen og virusmembranerne. Det ville ikke efterlade nogen energi til at trække membranerne sammen.
"Det er derfor, vi besluttede at lave en fuld beregning af systemet - alle proteinets atomer og alt vandet, " sagde Onuchic. "Det var en gigantisk indsats."
Den bevarede hydrofile (vandtiltrækkende) rest, kendt som Thr59, er af særlig interesse for forskerne, ikke kun for den måde, den forstyrrer foldning og tillader virussen at angribe, men også fordi den har en tvilling.
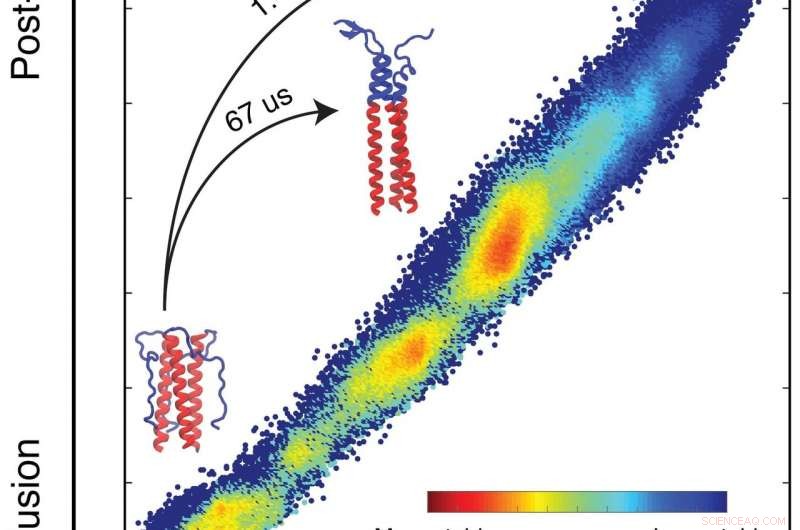
En simulering af biofysikere fra Rice University detaljerede den frie energiprofil, der dikterer, hvordan et protein, der hjælper influenzavirus med at inficere celler, udfører sin mission. Simuleringerne forudsiger, hvordan et protein vil foldes baseret på de iboende energier af hvert atom i systemet. Proteinerne danner sløjfer og spoler, når de søger deres laveste, mest stabile energitilstande (blå). På det område, forskerne undersøgte, de fandt et problem, der bremser foldningsprocessen, som gør det muligt at binde sig til målcellen, og som også giver nye vacciner mulighed for at angribe influenza. Klik på billedet for en større version. Kredit:Xingcheng Lin
"I det fulde evolutionære træ, disse vira falder i to grupper, og forskellen ser ud til at være denne rest, " sagde Onuchic. "De delte 1, 500 år siden og på en eller anden måde, efter denne adskillelse, de er fuldt bevarede. De har ikke været i stand til at ændre den rest, uanset hvad, og vi mener, at det gør denne rest vigtig."
Den nuværende forskning fokuserede på gruppen, der inkorporerer Thr59 og forårsager H3N2-stammen, der er ansvarlig for Hong Kong-influenzaen, sagde Lin. Den anden rest, Met59, optræder i H1N1-stammen, der forårsagede den spanske syge.
"Vi har stadig en lang vej at gå for at forstå hele proteinet, sagde han. Her, vi studerede kun et domæne af et protein, og der er flere andre, der er meget vigtige for dens funktion."
"Men det, Xingcheng allerede har gjort, er en beregningsmæssig tour de force, " tilføjede Onuchic. "Han viste, hvordan denne særlige rest bryder domænets spiralformede symmetri og gør det ustabilt nok til at give peptidet tid til at få fat i membranerne."
Sidste artikelCarboxylsyrer opfører sig som supersyrer på overfladen af vand
Næste artikelCannabidiol:Håb eller hype?
 Varme artikler
Varme artikler
-
 Biosensorteknologier til at tilbyde mere effektive tilgange til sygdomsbehandlingMonash Biomedicine Discovery Institutes professor Mibel Aguilar og Dr. John Lee ved siden af den nyudviklede biosensorteknologi. Kredit:Steve Morton Hver celle i vores kroppe er formet af dens y
Biosensorteknologier til at tilbyde mere effektive tilgange til sygdomsbehandlingMonash Biomedicine Discovery Institutes professor Mibel Aguilar og Dr. John Lee ved siden af den nyudviklede biosensorteknologi. Kredit:Steve Morton Hver celle i vores kroppe er formet af dens y -
 At få øje på det usynligeHøjopløsningsrøntgenstruktur af enzymet adenylatkinase fanget i en forbigående strukturel tilstand af en kovalent disulfidbinding. Kredit:Umeå Universitet Det er lykkedes kemikere ved Umeå Univers
At få øje på det usynligeHøjopløsningsrøntgenstruktur af enzymet adenylatkinase fanget i en forbigående strukturel tilstand af en kovalent disulfidbinding. Kredit:Umeå Universitet Det er lykkedes kemikere ved Umeå Univers -
 Molekylære tilsætningsstoffer forbedrer de mekaniske egenskaber af organisk solcellematerialeFremviser forskning fra professor GaneshBalasubramanians laboratorium (Group for Interface and Nanoengineering), Institut for Mekanik og Mekanik, Lehigh University, Bethlehem, USA. På trods af de sene
Molekylære tilsætningsstoffer forbedrer de mekaniske egenskaber af organisk solcellematerialeFremviser forskning fra professor GaneshBalasubramanians laboratorium (Group for Interface and Nanoengineering), Institut for Mekanik og Mekanik, Lehigh University, Bethlehem, USA. På trods af de sene -
 Fysikere opdager en tri-anion partikel med kolossal stabilitetKredit:Virginia Commonwealth University Forskere fra Virginia Commonwealth University har opnået en bedrift, der er den første inden for fysik og kemi-en der kunne have vidtrækkende anvendelser.
Fysikere opdager en tri-anion partikel med kolossal stabilitetKredit:Virginia Commonwealth University Forskere fra Virginia Commonwealth University har opnået en bedrift, der er den første inden for fysik og kemi-en der kunne have vidtrækkende anvendelser.
- Sådan finder du antallet af gram
- Årsag og effektvidenskabsprojekter
- 267 millioner mennesker verden over er i fare for havniveaustigning
- Solcellen, der også skinner:Selvlysende LED-design slår effektivitetsrekord
- Udrydde fejlene i klimamodeller for bedre at forudsige orkaner
- Sådan spore-test en autoklave