


Ny metode fremskynder simuleringer, giver ny indsigt i proteinfoldning
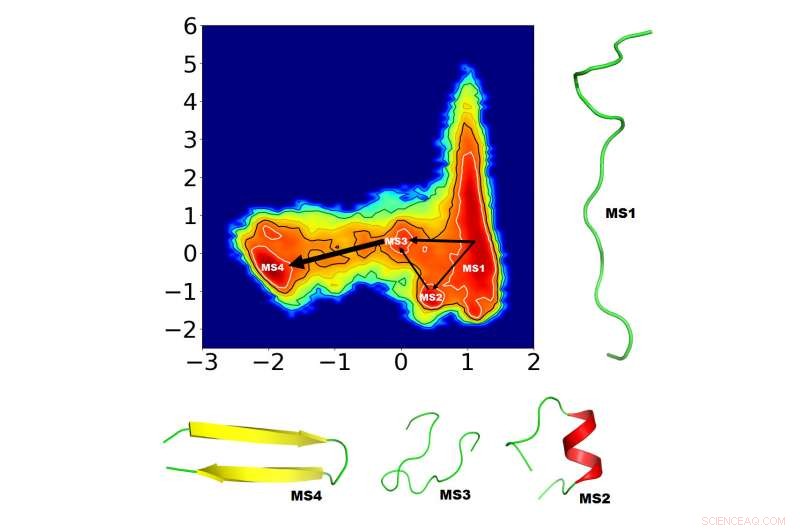
Forskere søger at forstå proteinfoldning bedre for at helbrede fejlfoldningssygdomme, men denne utroligt komplekse proces kræver sofistikerede algoritmer til at identificere foldemekanismerne. Beregningsbiofysikere har foreslået en ny måde at identificere de mest afgørende faktorer for proteinfoldning. De demonstrerede den korte simuleringstid af deres tilgang på et lille, men spændende protein, "GB1 beta-hårnål, "i The Journal of Chemical Physics . De fire nye mellemliggende foldningstilstande (MS1-4) identificeret af holdet er vist her, sammen med de mulige forbindelsesveje. Tykkelsen af de indbyrdes forbundne pile afspejler sandsynligheden for, at banen opstår. Kredit:Navjeet Ahalawat og Jagannath Mondal
Et proteins foldemønstre hjælper dem med at udføre deres dedikerede opgaver. Som cellens rigtige "gørere", selv en lille ændring i et proteins aminosyrerygrad kan forårsage fejlfoldning og hindre proteinets funktionalitet eller forårsage sygdom. For eksempel, hvis tau, et protein, der hjælper med at stabilisere strukturen af hjerneceller, er foldet forkert, det kan danne tau-tangles, som almindeligvis ses hos Alzheimers patienter.
Forskere søger at forstå proteinfoldning bedre for at helbrede fejlfoldningssygdomme, men denne utroligt komplekse proces kræver sofistikerede algoritmer til at identificere foldemekanismerne. Beregningsbiofysikere fra Tata Institute of Fundamental Research Hyderabad (TIFR-H) har foreslået en ny måde at identificere de mest afgørende faktorer for proteinfoldning. De demonstrerede den korte simuleringstid af deres tilgang på et lille, men spændende protein, "GB1 beta-hårnål, "i The Journal of Chemical Physics , fra AIP Publishing.
"Ved at kombinere en metode kendt som 'Tidsstrukturbaseret uafhængig komponentanalyse' (TICA) med korte molekylære dynamiksimuleringer, vi har fundet fire fysisk betydningsfulde mellemfoldningstilstande, ikke tidligere observeret, og viste spiralformede tilstande, som normalt ikke kan detekteres med andre metoder, " sagde Navjeet Ahalawat, en forfatter på papiret.
Hvert atom i et protein kan foldes i tre dimensioner, men med millioner af atomer til stede i selv simple proteiner, opgaven med at forstå den kollektive foldekombination bliver indviklet. Forskere har overvejet de forskellige faktorer, der påvirker proteinfoldning, såsom hydrogenbinding, og kombinerede disse til generelle beskrivelser kaldet kollektive variabler (CV'er). Imidlertid, med mange potentielle faktorer, forskere mangler en god måde at finde CV'er, der på passende vis beskriver en gennemførlig proces.
"Der er mange måder proteiner kan gå fra udfoldede til foldede tilstande, så det mest udfordrende er at beslutte, hvor man skal starte, " sagde Ahalawat. Jagannath Mondal, en anden forfatter på papiret, tilføjede, at det var let at "fare vild i dataene."
Holdet besluttede at studere den eksternt udragende hårnål af GB1-proteinet på grund af den store mængde eksisterende arbejde og mange potentielle foldningsmuligheder, der allerede er anslået i tidligere CV'er. Ahalawat og Mondal tog en række eksisterende GB1 CV'er som konstituerende CV'er og kombinerede dem lineært ved hjælp af TICA for at identificere et par "optimerede" CV'er. Derefter, de indlæste de optimerede CV'er i Markov State Model og identificerede fire mellemliggende foldningstilstande sammen med de mulige forbindelsesveje.
"Vi spurgte, hvad er de egenskaber, der tidligere er anslået for dette særlige protein, som virkelig kan spille en nøglerolle i systemet? Og kan vi finde den rigtige kombination af betingelser?" sagde Ahalawat. "I vores arbejde kan vi nu kvantitativt se, om den funktion overhovedet er relevant for processen."
"Ved brug af korte simuleringer, vi har fundet frem til den vægt, du virkelig skal bruge i en kombination, og dette giver det rigtige foldemønster for et protein, " tilføjede Mondal. "Det er en rigtig billig måde at finde ud af proteinfoldning på."
I deres metode, data fra tidligere undersøgelser er nødvendige for at identificere optimale CV'er. Holdet forestiller sig, at deres teknik kan bruges til at afdække den indre mekanisme af sund proteinfoldning for at korrigere sygdomsfremkaldende fejlfoldede proteiner. De ønsker også at videreudvikle deres CV-optimeringsmetode og anvende dem i biomolekylær genkendelse og lægemiddelopdagelse. "I fremtiden planlægger vi at inkorporere ikke-lineære metoder, ved at bruge neural-netværksbaserede deep learning-teknikker til at forbedre vores model, " sagde Ahalawat.
 Varme artikler
Varme artikler
-
 Hvilke brød formes hurtigere?De to mest forbrugte brødtyper i Amerika er hvide og multigrain. Nogle familier kan vælge at bage deres eget brød, andre foretrækker måske at købe økologisk brød, men hver familie ved, at hvis de ikke
Hvilke brød formes hurtigere?De to mest forbrugte brødtyper i Amerika er hvide og multigrain. Nogle familier kan vælge at bage deres eget brød, andre foretrækker måske at købe økologisk brød, men hver familie ved, at hvis de ikke -
 Hvad er forskellen mellem relativ luftfugtighed og dugpunkt?Kredit:The American Chemical Society Meteorologer rapporterer ofte mængden af fugt i luften som relativ fugtighed eller dugpunkt. Disse foranstaltninger kan være forvirrende for folk, der bare
Hvad er forskellen mellem relativ luftfugtighed og dugpunkt?Kredit:The American Chemical Society Meteorologer rapporterer ofte mængden af fugt i luften som relativ fugtighed eller dugpunkt. Disse foranstaltninger kan være forvirrende for folk, der bare -
 Producerer grønt brint ved udsættelse af nanomaterialer for sollysSe gennem et vindue af det indre af en ultrahøj vakuumreaktor, hvor TiO2-nanorør er dekoreret med CoO-nanopartikler. Vi ser flammen (plasma produceret ved laserablation), der sprutter CoOet for at giv
Producerer grønt brint ved udsættelse af nanomaterialer for sollysSe gennem et vindue af det indre af en ultrahøj vakuumreaktor, hvor TiO2-nanorør er dekoreret med CoO-nanopartikler. Vi ser flammen (plasma produceret ved laserablation), der sprutter CoOet for at giv -
 Ingredienser i Carburetor CleanersCarburetor Cleaners er enten aerosoler med en dåse eller findes i portioner i gallon-størrelse. Toksiciteten af en forgasserrensers hovedbestanddele gør i sig selv denne cocktail til et farligt stof
Ingredienser i Carburetor CleanersCarburetor Cleaners er enten aerosoler med en dåse eller findes i portioner i gallon-størrelse. Toksiciteten af en forgasserrensers hovedbestanddele gør i sig selv denne cocktail til et farligt stof
- Bæredygtighedspåstande om gummi klæber ikke
- Hvorfor studerer forskere fossiler?
- Fysikere viser, at det er umuligt at maskere kvanteinformation i korrelationer
- Jordens magnetiske poler kan begynde at vende. Hvad sker der så?
- Kemikere rapporterer en detaljeret beskrivelse af azopyrazoloniske farvestoffer
- Eksperter behandler måder, hvorpå de kan understøtte de nyeste standarder for naturvidenskabelig …