


Evolutionær koblingsanalyse identificerer virkningen af sygdomsassocierede varianter
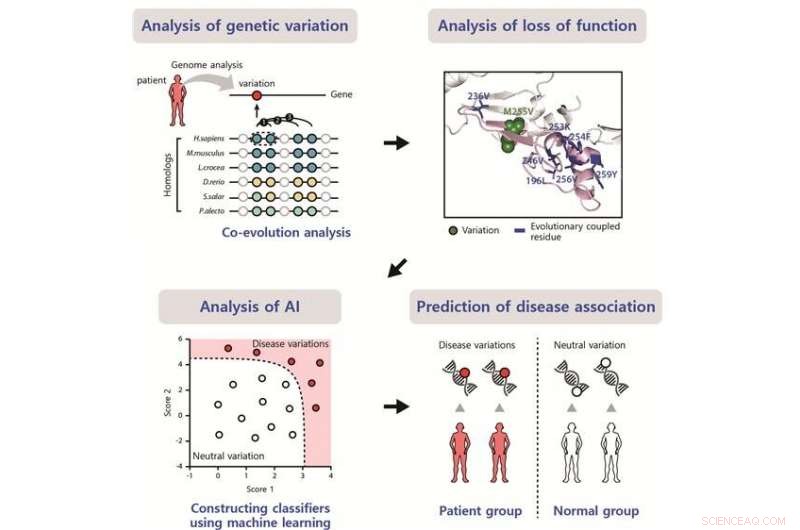
Skematisk af den udviklede metode til at identificere virkningen af sygdomsassocierede varianter. Kredit:POSTECH
Forudsigelse af virkningen af DNA-sekvensvarianter er vigtig for sortering af sygdomsassocierede varianter (DV'er) fra neutrale varianter. Koreanske forskere ved Pohang University of science and technology (POSTECH) rapporterer udviklingen af en metode til at forudsige virkningen af DVs. Undersøgelsen vises i tidsskriftet Nukleinsyreforskning i juni.
Nuværende metoder til at forudsige de mutationelle virkninger afhænger af evolutionær bevarelse på mutationsstedet, som bestemmes ved hjælp af homologe sekvenser og baseret på den antagelse, at varianter på velkonserverede steder har høje virkninger. Imidlertid, mange DV'er på mindre bevarede, men funktionelt vigtige steder, kan ikke forudsiges af de nuværende metoder.
Forskerne præsenterer en metode til at finde DV'er på mindre bevarede steder ved at forudsige de mutationelle virkninger ved hjælp af evolutionær koblingsanalyse. Funktionelt vigtige og evolutionært koblede steder har ofte kompenserende varianter på samarbejdssteder for at undgå funktionstab. De identificerede DV'er på mindre konserverede steder, der ikke blev identificeret ved hjælp af nuværende konserveringsbaserede metoder.
Prof. Kim sagde, "Denne undersøgelse kan anvendes på en række præcisionsmedicinske metoder, såsom prognose for patientens sygdomme og finde personlig medicin. Baseret på en stor skala sekvensanalyse, den udviklede metode er nyttig til at finde flere sygdomsassocierede varianter, som hjælper med at finde biomarkører og terapeutiske mål for forskellige menneskelige sygdomme."
Sidste artikelLadningsoverførsel inden for overgangsmetalfarvestoffer analyseret
Næste artikelOrganiske solceller, der holder 10 år i rummet
 Varme artikler
Varme artikler
-
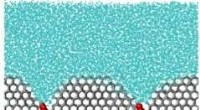 Polymerer til undsætning! Redder celler fra at beskadige isEn simulering af et ishæmmende molekyle. Molekylet, i rødt, er som en vægt på overfladen af iskrystallen, krum den og forhindrer yderligere iskrystalvækst. Kredit:University of Utah Celleterapie
Polymerer til undsætning! Redder celler fra at beskadige isEn simulering af et ishæmmende molekyle. Molekylet, i rødt, er som en vægt på overfladen af iskrystallen, krum den og forhindrer yderligere iskrystalvækst. Kredit:University of Utah Celleterapie -
 Glas fra en 3-D printerForskellige glasobjekter lavet med en 3-D printer. Kredit:Group for Complex Materials / ETH Zürich ETH-forskere brugte en 3-D printproces til at fremstille komplekse og meget porøse glasobjekter.
Glas fra en 3-D printerForskellige glasobjekter lavet med en 3-D printer. Kredit:Group for Complex Materials / ETH Zürich ETH-forskere brugte en 3-D printproces til at fremstille komplekse og meget porøse glasobjekter. -
 Båndets historie:Klæbrige bits gør bedre batterierTil venstre, en kobberstrømsamler med en laserinduceret siliciumoxidbelægning skabt på Rice University. Til højre, et scanningselektronmikroskopbillede af belægningen skabt ved lasning af tape på kobb
Båndets historie:Klæbrige bits gør bedre batterierTil venstre, en kobberstrømsamler med en laserinduceret siliciumoxidbelægning skabt på Rice University. Til højre, et scanningselektronmikroskopbillede af belægningen skabt ved lasning af tape på kobb -
 Fyrrenåle fra gamle juletræer kan i fremtiden blive til maling og sødemidlerKredit:CC0 Public Domain Forladte juletræer kunne reddes fra losseplads og omdannes til maling og sødemidler til mad ifølge ny forskning fra University of Sheffield. Juletræer har hundredtusindvi
Fyrrenåle fra gamle juletræer kan i fremtiden blive til maling og sødemidlerKredit:CC0 Public Domain Forladte juletræer kunne reddes fra losseplads og omdannes til maling og sødemidler til mad ifølge ny forskning fra University of Sheffield. Juletræer har hundredtusindvi
- Metalfri katalysator udvider rækkevidden af estersyntese
- Jordsundhed er lige så vigtigt for miljøet som luft- og vandkvalitet, siger mikrobiologer
- Tab af individualitet på grund af genetisk teknik
- Bil dele, skistøvler og kasser:Hvordan ødelagt eller brugt plastik får nyt liv
- Weyl-Kondo semimetal:Fysikere opdager en ny type kvantemateriale
- Neutroner undersøger tomater for at få indsigt i interplant-snak