


Molekylær dynamiksimulering kaster nyt lys over metanhydratdannelse

Methanhydrat hentet fra havbunden ud for Oregons kyst, USA. Kredit:Wikimedia Commons
I et papir, der blev offentliggjort i denne uge i PNAS , forskere ved University of Amsterdams Van 't Hoff Institute for Molecular Sciences og Amsterdam Center for Multiscale Modeling giver atomistisk indsigt i dannelsen af metanhydrater. På grundlag af molekylære dynamiksimuleringer forklarer de, hvordan valg mellem konkurrerende methanhydratpolymorfer sker, og hvordan dette kan generaliseres til andre hydrater og molekylær krystaldannelse.
Methanhydrat er islignende faste stoffer, der er rigeligt til stede, blandt andet ved havbunden. Det anslås, at mængden af energi, der er lagret i methanhydrater, er dobbelt så stor som den mængde energi, der er lagret i konventionelle fossile brændstoffer. På samme tid, dannelsen af hydrater bekymrer olieindustrien, da de kan tilstoppe olierørledninger, forårsager flowproblemer. Methanhydrater er også til stede i permafrosten i arktiske områder. Optøning af permafrosten som følge af stigende globale temperaturer kan føre til frigivelse af store mængder metan, som er en kraftig drivhusgas.
Kemiske metanmolekyler
I et metanhydrat, på molekylært niveau er metan indkapslet inde i et hydrogenbundet vandnetværk. Mens metangas er hydrofob under omgivelsesbetingelser, ved lave temperaturer og høje tryk kan en blanding af vand og metangas spontant nukleere til hydrater.
I årenes løb, interessen for at forstå hydraters dannelsesmekanisme er steget enormt. Især deres dannelse under naturlige forhold er dårligt forstået. Forståelse af processen med homogen nukleation, og hvordan dette fører til forskellige metanhydratpolymorfer, kan føre til forbedret kontrol af krystallisation, samt indsigt i polymorfvalg generelt.
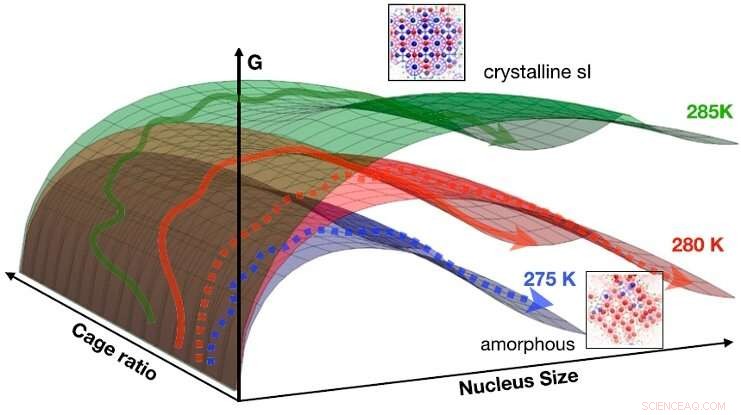
Resultaterne kan opsummeres i en idealiseret CNT fri energioverflade som funktion af størrelse og burforhold for 275 K (blå), 280 K (rød), og 285 K (grøn). Pilene angiver skematisk veje, der krydser fra væske til fast stof (stiplede pile:til amorf fase; faste pile:til krystallinsk fase). Ved lave temperaturer (f.eks. 275 K), fri energibarrieren for at nukleere det amorfe faste stof er lavest, tendensen er vendt ved højere temperatur (f.eks. 285 K), hvor prøvetagningsveje for det meste ender i den krystallinske fase. Ved 280 K er begge mekanismer tilgængelige. Kredit:HIMS/PNAS
Ny tilgang til simulering
Da eksperimentel forskning om dannelsen af de forskellige methanhydratpolymorfer lider under begrænset opløsning, Amsterdam -forskerne under ledelse af professor Peter Bolhuis brugte molekylære dynamiksimuleringer for at give sådan indsigt.
Anvendelse af en direkte molekylær dynamiksimulering er ikke særlig effektiv, fordi ved moderat afkøling er nukleation en meget sjælden hændelse, på grund af tilstedeværelsen en meget høj energibarriere. En sådan simulering ville kræve beregningstider ud over universets alder. Imidlertid, fordi kernehændelsen i sig selv, mens det er sjældent, sker meget hurtigt (på en mikrosekund tidsskala), forskerne kunne oprette en stor samling af molekylære dynamikbaner, der udviser disse hurtige begivenheder. Efterfølgende detaljeret analyse af disse baner viste, hvordan selektion mellem konkurrerende amorfe og krystallinske polymorfe dannelsesmekanismer finder sted. Deres PNAS -papir kaster ikke kun lys over dannelsen af metanhydrater, men også på andre clathratforbindelser og molekylær krystaldannelse generelt.
 Varme artikler
Varme artikler
-
 2D-materialer tilbyder unikke strækegenskaberAuxetiske materialer bliver tykkere, når de strækkes og tyndere, når de komprimeres. Det auxetiske materiale til venstre vokser i bredden, når det trækkes i lodret retning, som vist til højre. Kredit:
2D-materialer tilbyder unikke strækegenskaberAuxetiske materialer bliver tykkere, når de strækkes og tyndere, når de komprimeres. Det auxetiske materiale til venstre vokser i bredden, når det trækkes i lodret retning, som vist til højre. Kredit: -
 Nye forskningsresultater kan føre til sikrere og mere kraftfulde lithium-ion-batterierKredit:CC0 Public Domain Virginia Commonwealth University-forskere arbejder på at forbedre ledningsevne og sikkerhed i lithium-ion-batterier, som bruges til at drive mange elektroniske enheder run
Nye forskningsresultater kan føre til sikrere og mere kraftfulde lithium-ion-batterierKredit:CC0 Public Domain Virginia Commonwealth University-forskere arbejder på at forbedre ledningsevne og sikkerhed i lithium-ion-batterier, som bruges til at drive mange elektroniske enheder run -
 De udvidede muligheder for biobaserede polymererKredit:Institut for Kemisk Forskning i Catalonien At finde innovative og bæredygtige løsninger til vores materialebehov er et af kernemålene for grøn kemi. De utallige plastik, der omslutter vores
De udvidede muligheder for biobaserede polymererKredit:Institut for Kemisk Forskning i Catalonien At finde innovative og bæredygtige løsninger til vores materialebehov er et af kernemålene for grøn kemi. De utallige plastik, der omslutter vores -
 Mikrofluidisk array fanger, holder enkelte livmoderhalsceller for hurtigere screeningEksperimentelle resultater af immunfarvede celler under anvendelse af mikrobrøndsarrayet med barrierer. Kredit:Soo Hyeon Kim Der er udviklet flere screeningstest for livmoderhalskræft i de senere
Mikrofluidisk array fanger, holder enkelte livmoderhalsceller for hurtigere screeningEksperimentelle resultater af immunfarvede celler under anvendelse af mikrobrøndsarrayet med barrierer. Kredit:Soo Hyeon Kim Der er udviklet flere screeningstest for livmoderhalskræft i de senere
- Drysset med kraft:Hvordan urenheder forstærker et termoelektrisk materiale på atomniveau
- Autonomt køretøj for at forbedre integrerede transportløsninger
- Additiv fremstilling af cellulosebaserede materialer med kontinuerlig, stigninger i flere retninger
- Forskere finder beviser for trafikforurening i det fjerne Himalaya
- Kvarterets trivsel og fællesskabsfølelse er kernen i et godt hjem, siger forskere
- Fokker Dr I Triplane