


Forskere producerer de første open source-all-atom-modeller af COVID-19 spike protein
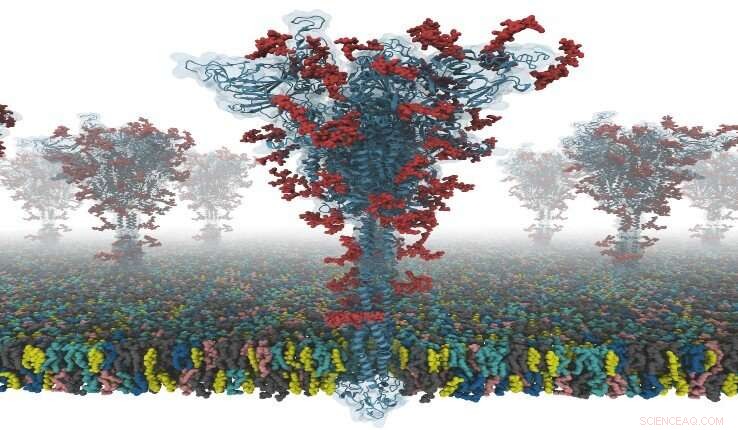
En model af et S-protein. Kredit:Dr. Yeolkyo Choi/Lehigh
Virus SARS coronavirus 2 (SARS-CoV-2) er den kendte årsag til coronavirus sygdom 2019 (COVID-19). "Spike"- eller S-proteinet letter viral indtræden i værtsceller.
Nu er en gruppe forskere fra Seoul National University i Sydkorea, University of Cambridge i Storbritannien, og Lehigh University i USA, har arbejdet sammen om at producere de første open source alle-atom-modeller af et fuldlængde S-protein. Forskerne siger, at dette er af særlig betydning, fordi S-proteinet spiller en central rolle i viral indtræden i celler, gør det til et hovedmål for udvikling af vacciner og antivirale lægemidler.
Detaljerne kan findes i et papir, "Udvikling af en fuldt glykosyleret fuldlængde SARS-CoV-2 Spike Protein Model in a Viral Membrane" netop offentliggjort online i Journal of Physical Chemistry B .
Denne videodemo illustrerer, hvordan man bygger dette membransystem ud fra deres SARS-CoV-2 S-proteinmodeller. Modelbygningsprogrammet er åbent og kan findes fra CHARMM-GUIs hjemmeside ved at klikke på COVID-19-arkivlinket, eller ved at klikke på arkivlinket i overskriften, derefter COVID-19 Proteins-linket i venstre sidebjælke.
Udviklet af Wonpil Im, en professor i Lehigh University's Institut for Biologiske Videnskaber og Bioingeniørafdeling, CHARMM-GUI (GUI =grafisk brugergrænseflade) er et program, der simulerer komplekse biomolekylære systemer ganske enkelt, præcist og hurtigt. Jeg beskriver det som et "beregningsmikroskop", der gør det muligt for forskere at forstå interaktioner på molekylært niveau, som ikke kan observeres på anden måde. Mere information om CHARMM-GUI kan findes i denne video.
"Vores modeller er de første fuldt glykosylerede fuldlængde SARS-CoV-2 spike (S) proteinmodeller, der er tilgængelige for andre forskere, " siger Im. "Jeg var så heldig at samarbejde med Dr. Chaok Seok fra Seoul National University i Korea og Dr. Tristan Croll fra University of Cambridge i Storbritannien. Vores team brugte dage og nætter på at bygge disse modeller meget omhyggeligt ud fra den kendte kryo- EM struktur dele. Modellering var meget udfordrende, fordi der var mange regioner, hvor simpel modellering ikke kunne levere modeller af høj kvalitet."
Forskere kan bruge modellerne til at udføre innovativ og ny simuleringsforskning til forebyggelse og behandling af COVID-19, ifølge Im.
S-proteinstrukturen blev bestemt med cryo-EM med RBD op (PDB ID:6VSB), og med RBD nede (PDB ID:6VXX). Men, denne model har mange manglende rester. Så, de modellerede først de manglende aminosyrerester, og så andre manglende domæner. Ud over, de modellerede alle potentielle glycaner (eller kulhydrater) knyttet til S-proteinet. Disse glykaner forhindrer antistofgenkendelse, hvilket gør det svært at udvikle en vaccine. De byggede også et viralt membransystem af et S-protein til simulering af molekylær dynamik.
 Varme artikler
Varme artikler
-
 Ny bøjelig smartphone-teknologi kan bruge overvågning til at redde patienters livPurdue University-forskere arbejder på at bruge en ny polymerfilm, som kunne gøre smartphones mere bøjelige, at skabe skræddersyede sensorer, der ikke-invasivt kunne overvåge glukoseniveauer, hjertefr
Ny bøjelig smartphone-teknologi kan bruge overvågning til at redde patienters livPurdue University-forskere arbejder på at bruge en ny polymerfilm, som kunne gøre smartphones mere bøjelige, at skabe skræddersyede sensorer, der ikke-invasivt kunne overvåge glukoseniveauer, hjertefr -
 Elektroner øger faststof kaloriekøling i hexagonale sulfiderEt skitsekort for mekanismen for kæmpe barokalorisk (BC) effekt i Ni1-xFexS (op mønster). En sammenligning af volumennormaliseret entropiændring drevet af 100 MPa og termisk ledningsevne (k) for Ni 0
Elektroner øger faststof kaloriekøling i hexagonale sulfiderEt skitsekort for mekanismen for kæmpe barokalorisk (BC) effekt i Ni1-xFexS (op mønster). En sammenligning af volumennormaliseret entropiændring drevet af 100 MPa og termisk ledningsevne (k) for Ni 0 -
 Ny metode til at fremstille kompositter med formhukommelseFormhukommelsestest af hærdet harpiks og pultruderede kompositprøver:(a) Deformeret prøve af hærdet harpiks i testfiksturen efter opvarmning; (b) hærdede harpiksprøver efter formfiksering; (c) hærdede
Ny metode til at fremstille kompositter med formhukommelseFormhukommelsestest af hærdet harpiks og pultruderede kompositprøver:(a) Deformeret prøve af hærdet harpiks i testfiksturen efter opvarmning; (b) hærdede harpiksprøver efter formfiksering; (c) hærdede -
 Tilføjelse af en inert polymer til plastsolceller muliggør høj effektivitet og let produktionTilføjelse af en inert polymerkomponent i plastsolceller -som visualiseret ved pilene i dette billede -skaber en unik søjleform og muliggør øget optimal enhedstykkelse, som er meget bedre egnet til in
Tilføjelse af en inert polymer til plastsolceller muliggør høj effektivitet og let produktionTilføjelse af en inert polymerkomponent i plastsolceller -som visualiseret ved pilene i dette billede -skaber en unik søjleform og muliggør øget optimal enhedstykkelse, som er meget bedre egnet til in
- Mysteriet om neutronens levetid
- Nanoskalaudvikling øger ydeevnen for quantum dot lysemitterende dioder
- Selv tekniske ledere bekymrer sig om deres børns smartphone-afhængighed
- Demonstration af kvantekommunikation over optiske fibre over 600 km
- Hvad en ubelejlig sandhed fik ret (og forkert) om klimaændringer
- Hvad sker der, når du skærer en magnet i halvdelen?