


Gennembrud rapporteret inden for maskinlæringsforbedret kvantekemi
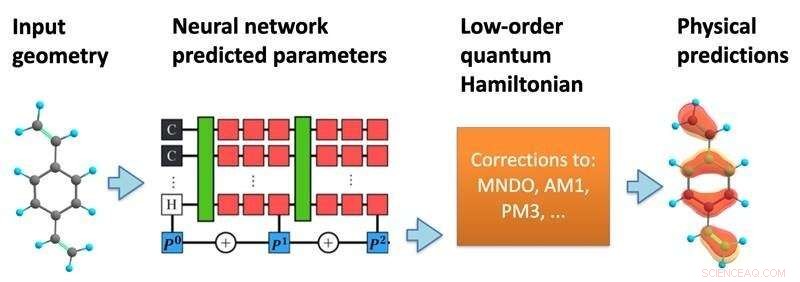
Modellens opbygning. Et neuralt netværk behandler en molekylær geometri for at forudsige en semi-empirisk kvante Hamiltonian, som derefter løses selvkonsekvent for at forudsige en række kemiske egenskaber. Kredit:Kipton Barros, Los Alamos National Laboratory.
I en ny undersøgelse, offentliggjort i Proceedings of the National Academy of Sciences , har forskere fra Los Alamos National Laboratory foreslået at inkorporere mere af kvantemekanikkens matematik i strukturen af maskinlæringsforudsigelserne. Ved at bruge de specifikke positioner af atomer i et molekyle forudsiger maskinlæringsmodellen en effektiv Hamiltonian matrix, som beskriver de forskellige mulige elektroniske tilstande sammen med deres tilknyttede energier.
Sammenlignet med traditionelle kvantekemi-simuleringer laver den maskinlæringsbaserede tilgang forudsigelser til en meget reduceret beregningsomkostning. Det muliggør kvantitativt præcise forudsigelser vedrørende materialeegenskaber, tillader fortolkelig indsigt i arten af kemisk binding mellem atomer og kan bruges til at forudsige andre komplekse fænomener, såsom hvordan systemet vil reagere på forstyrrelser, såsom lys-stof-interaktioner. Metoden giver også meget forbedret nøjagtighed i forhold til traditionelle maskinlæringsmodeller og demonstrerer succes i overførbarhed, dvs. modellens evne til at lave forudsigelser, der går langt ud over de data, der dannede grundlaget for dens træning.
Kvantemekanikkens ligninger giver en køreplan til at forudsige kemikaliers egenskaber ud fra grundlæggende videnskabelige teorier. Disse ligninger kan dog hurtigt blive for dyre i form af computertid og strøm, når de bruges til at forudsige adfærd i store systemer. Machine learning tilbyder en lovende tilgang til at accelerere sådanne simuleringer i stor skala. Brugen af maskinlæring til at forudsige kemiske egenskaber rummer potentialet for store teknologiske fremskridt, med anvendelser fra renere energi til hurtigere farmaceutiske lægemiddeldesign. Dette er et meget aktivt forskningsområde, men de fleste eksisterende tilgange bruger simple og heuristiske tilgange til design af maskinlæringsmodellerne.
I deres undersøgelse har forskerne vist, at maskinlæringsmodeller kan efterligne grundstrukturen i de grundlæggende naturlove. Disse love kan være meget svære at simulere direkte. Maskinlæringstilgangen muliggør forudsigelser, der er nemme at beregne og er nøjagtige i en lang række kemiske systemer.
Den forbedrede maskinlæringsmodel kan hurtigt og præcist forudsige en lang række egenskaber ved molekyler. Disse tilgange scorer meget godt på vigtige benchmarks inden for beregningskemi og viser, hvordan deep learning-metoder fortsat kan forbedres ved at inkorporere flere data fra eksperimenter. Modellen kan også lykkes med udfordrende opgaver som at forudsige ophidsede tilstandsdynamikker - hvordan systemer opfører sig med forhøjede energiniveauer. Dette værktøj er en banebrydende evne til kvantekemi. Det vil give forskere mulighed for bedre at forstå nye molekylers reaktivitet og exciterede tilstande. + Udforsk yderligere
Computere udmærker sig i kemiklassen
 Varme artikler
Varme artikler
-
 En ny strategi til syntetisering af 2-D uorganiske materialer, der bruges i kondensatorer, batterier…Overfladereaktioner af MXener i smeltede uorganiske salte. (A) Skemaer for ætsning af MAX faser i Lewis sure smeltede salte. (B) Atomisk opløsning højvinklet ringformet mørkt felt (HAADF) billede af T
En ny strategi til syntetisering af 2-D uorganiske materialer, der bruges i kondensatorer, batterier…Overfladereaktioner af MXener i smeltede uorganiske salte. (A) Skemaer for ætsning af MAX faser i Lewis sure smeltede salte. (B) Atomisk opløsning højvinklet ringformet mørkt felt (HAADF) billede af T -
 En miljøvenlig teknik til at opgradere metalaffald til multifunktionelle aerogelerDisse metalbaserede aerogeler har høj termisk og mekanisk stabilitet, og de kunne potentielt bruges som lette byggematerialer og til dyrkning af celler til biomedicinske formål. Kredit:National Univer
En miljøvenlig teknik til at opgradere metalaffald til multifunktionelle aerogelerDisse metalbaserede aerogeler har høj termisk og mekanisk stabilitet, og de kunne potentielt bruges som lette byggematerialer og til dyrkning af celler til biomedicinske formål. Kredit:National Univer -
 Hvordan bittesmå enzymer troner øverst i verdensomspændende kulstofgenbrugHvide rådsvampe, Duke Forest, North Carolina Kredit:NA Genanvendelsen af det meste af kulstoffet i naturen afhænger af nedbrydning af to polymerer i træemateriale, især cellulose og lignin. I et
Hvordan bittesmå enzymer troner øverst i verdensomspændende kulstofgenbrugHvide rådsvampe, Duke Forest, North Carolina Kredit:NA Genanvendelsen af det meste af kulstoffet i naturen afhænger af nedbrydning af to polymerer i træemateriale, især cellulose og lignin. I et -
 Forbedret omgivende ammoniakfotosyntese ved hjælp af nanosheets med lysomskiftelige iltpladserForskere har præsenteret en strategi ved samtidig at introducere lysomskiftelig ilt-ledighed og dopingmo i Bi 5 O 7 Br nanosheets til effektivt fotokatalytisk N 2 fiksering. Den modificerede fot
Forbedret omgivende ammoniakfotosyntese ved hjælp af nanosheets med lysomskiftelige iltpladserForskere har præsenteret en strategi ved samtidig at introducere lysomskiftelig ilt-ledighed og dopingmo i Bi 5 O 7 Br nanosheets til effektivt fotokatalytisk N 2 fiksering. Den modificerede fot