


En model trænet til at forudsige spektroskopiske profiler hjælper med at tyde strukturen af materialer
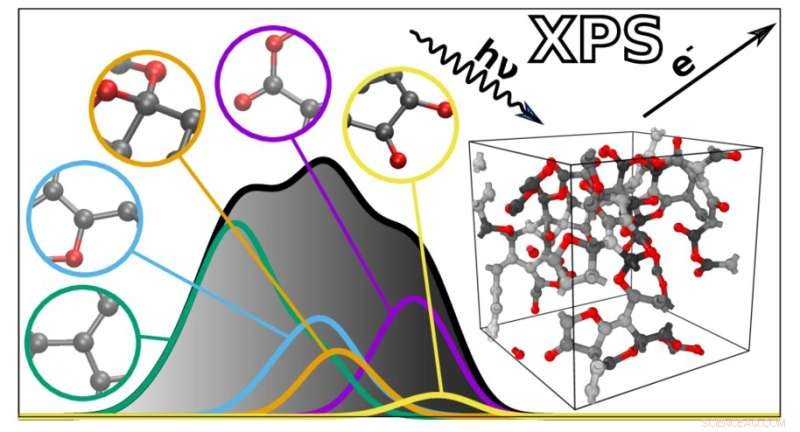
Den nye algoritme forudsiger XPS-spektrene for komplekse materialer baseret på individuelle atomare bidrag. Kredit:Miguel Caro / Aalto University
Kulstofbaserede materialer rummer et enormt potentiale for at bygge en bæredygtig fremtid, men materialeforskere har brug for værktøjer til korrekt at analysere deres atomare struktur, som bestemmer deres funktionelle egenskaber. Røntgenfotoelektronspektroskopi (XPS) er et af de værktøjer, der bruges til at gøre dette, men XPS-resultater kan være udfordrende at fortolke. Nu har forskere hos Aalto udviklet et maskinlæringsværktøj til at forbedre XPS-analyser, som de har gjort frit tilgængeligt som XPS Prediction Server.
XPS-spektre er grafer med en samling af toppe, der afspejler bindingsenergien af elektronerne dybt inde i de atomer, der udgør et materiale. Fordi bindingsenergierne afhænger af det atomare miljø, kan de bruges til at udlede, hvordan atomerne er forbundet i et bestemt materiale eller molekyle. Dette gør dog også XPS-spektre svære at fortolke, da mange faktorer påvirker bindingsenergierne. Bindingsenergierne af forskellige atomare egenskaber kan også overlappe hinanden, hvilket yderligere komplicerer analysen.
For at hjælpe med dette udviklede et team ledet af Miguel Caro en beregningsmetode, der kan forudsige det bindende energispektrum af et materiale baseret på en computergenereret strukturel model. Dette forenkler XPS-datafortolkning ved at gøre det muligt at matche de eksperimentelt observerede bindingsenergier mod de beregningsmæssige forudsigelser.
Ideen i sig selv er ikke ny, men problemet har været den beregningsmæssige vanskelighed med at beregne XPS-spektret af et materiale nøjagtigt. Caros team løste dette ved hjælp af maskinlæring. Tricket var at træne en billig computeralgoritme til at forudsige resultatet af en beregningsmæssigt dyr referencemetode baseret på en effektiv kombination af beregningsmæssigt billige og dyre kvantemekaniske data.
Den beregningsmæssigt billigere metode, DFT, matcher ikke eksperimentelle resultater særlig præcist. Den mere nøjagtige metode, GW, tager for lang tid at beregne, når et molekyle har mange atomer. "Vi besluttede at konstruere en basismodel, der bruger rigelige DFT-data og derefter forfine den med sparsomme og dyrebare GW-data. Og det virkede," siger Caro.
Den resulterende algoritme kan forudsige spektret af ethvert uordnet materiale lavet af kulstof, brint og oxygen. "De forudsagte spektre er bemærkelsesværdigt tæt på dem, der opnås eksperimentelt. Dette åbner døren til bedre integration mellem eksperimentel og beregningsmæssig karakterisering af materialer," siger Caro. Dernæst planlægger holdet at udvide deres teknik til at omfatte et bredere udvalg af materialer og andre typer spektroskopi.
Open access-artiklen blev publiceret i Chemistry of Materials . + Udforsk yderligere
Diamantlignende kulstof dannes anderledes end man troede – maskinlæring muliggør udvikling af ny model
 Varme artikler
Varme artikler
-
 Bro over koblede farvande:Forskere 3-D-printer laboratorium med fuld væske på en chipNår to væsker - den ene indeholder nanoskala lerpartikler, en anden indeholdende polymerpartikler - er trykt på et glassubstrat, de kommer sammen ved grænsefladen mellem de to væsker og danner inden f
Bro over koblede farvande:Forskere 3-D-printer laboratorium med fuld væske på en chipNår to væsker - den ene indeholder nanoskala lerpartikler, en anden indeholdende polymerpartikler - er trykt på et glassubstrat, de kommer sammen ved grænsefladen mellem de to væsker og danner inden f -
 Forskere skaber slangegift-afledt superlim, der stopper blødning på få sekunder ved hjælp af syn…Bothrops atrox. Kredit:Bernard Dupont, Wikimedia Commons Indiana Jones hader slanger. Og han er bestemt ikke alene. Frygten for slanger er så almindelig, at den endda har sit eget navn:ophidiophob
Forskere skaber slangegift-afledt superlim, der stopper blødning på få sekunder ved hjælp af syn…Bothrops atrox. Kredit:Bernard Dupont, Wikimedia Commons Indiana Jones hader slanger. Og han er bestemt ikke alene. Frygten for slanger er så almindelig, at den endda har sit eget navn:ophidiophob -
 Et nyt mikroskop afslører miraklet ved molekylær iltKunstners indtryk af interaktionen mellem tripletilstanden (blå pile) af et individuelt pentacenmolekyle (sort og hvidt) med et iltmolekyle (rødt). Kredit:Jascha Repp Forskere ved University of Re
Et nyt mikroskop afslører miraklet ved molekylær iltKunstners indtryk af interaktionen mellem tripletilstanden (blå pile) af et individuelt pentacenmolekyle (sort og hvidt) med et iltmolekyle (rødt). Kredit:Jascha Repp Forskere ved University of Re -
 Softwareprogrammet Allchemy identificerer præbiotisk syntese af biokemiske forbindelser fra urpræk…Et team af forskere, der arbejder i Institut for Organisk Kemis laboratorium på det polske videnskabsakademi, har udviklet et softwareprodukt til at hjælpe med at opdage de kemiske processer, der ført
Softwareprogrammet Allchemy identificerer præbiotisk syntese af biokemiske forbindelser fra urpræk…Et team af forskere, der arbejder i Institut for Organisk Kemis laboratorium på det polske videnskabsakademi, har udviklet et softwareprodukt til at hjælpe med at opdage de kemiske processer, der ført
- Billede:X-ray øje af Athena
- Hvordan en global havtraktat kunne beskytte biodiversiteten i det åbne hav
- De to styrker, der holder planeterne i bevægelse omkring solen
- Vind af rubiner og safirer rammer himlen på den gigantiske planet
- Rocket Lab siger, at jordudstyr skæmmede New Zealands opsendelse
- Air Canada genansætter 16, 500 arbejdere afskediget på grund af pandemi