


AI designer aktive farmaceutiske ingredienser hurtigt og nemt baseret på proteinstrukturer

En ny computerproces udviklet af kemikere ved ETH Zürich gør det muligt at generere aktive farmaceutiske ingredienser hurtigt og nemt baseret på et proteins tredimensionelle overflade. Den nye proces, beskrevet i Nature Communications , kunne revolutionere lægemiddelforskningen.
"Det er et reelt gennembrud for lægemiddelopdagelse," siger Gisbert Schneider, professor ved ETH Zürichs afdeling for kemi og anvendt biovidenskab. Sammen med sin tidligere doktorand Kenneth Atz har han udviklet en algoritme, der bruger kunstig intelligens (AI) til at designe nye aktive farmaceutiske ingredienser.
For ethvert protein med en kendt tredimensionel form genererer algoritmen tegningerne for potentielle lægemiddelmolekyler, der øger eller hæmmer proteinets aktivitet. Kemikere kan derefter syntetisere og teste disse molekyler i laboratoriet.
Alt hvad algoritmen behøver er et proteins tredimensionelle overfladestruktur. Baseret på det designer den molekyler, der binder specifikt til proteinet i henhold til lås og nøgle-princippet, så de kan interagere med det.
Ekskluderer bivirkninger fra starten
Den nye metode bygger på kemikeres årtier lange bestræbelser på at belyse proteiners tredimensionelle struktur og bruge computere til at søge efter egnede potentielle lægemiddelmolekyler. Indtil nu har dette ofte involveret besværligt manuelt arbejde, og i mange tilfælde har eftersøgningen givet molekyler, som var meget svære eller umulige at syntetisere. Hvis forskere overhovedet har brugt AI i denne proces i de senere år, var det primært for at forbedre eksisterende molekyler.
Nu, uden menneskelig indgriben, er en generativ AI i stand til at udvikle lægemiddelmolekyler fra bunden, der matcher en proteinstruktur. Denne banebrydende nye proces sikrer lige fra starten, at molekylerne kan syntetiseres kemisk. Derudover foreslår algoritmen kun molekyler, der interagerer med det specificerede protein på det ønskede sted og næsten ikke med andre proteiner.
"Det betyder, at når vi designer et lægemiddelmolekyle, kan vi være sikre på, at det har så få bivirkninger som muligt," siger Atz.
For at skabe algoritmen trænede forskerne en AI-model med information fra hundredtusindvis af kendte interaktioner mellem kemiske molekyler og de tilsvarende tredimensionelle proteinstrukturer.
Succesfulde tests med industrien
Sammen med forskere fra medicinalvirksomheden Roche og andre samarbejdspartnere testede ETH-teamet den nye proces og demonstrerede, hvad den er i stand til.
Forskerne søgte efter molekyler, der interagerer med proteiner i PPAR-klassen - proteiner, der regulerer sukker- og fedtsyremetabolismen i kroppen. Adskillige diabetesmedicin, der bruges i dag, øger aktiviteten af PPAR'er, hvilket får cellerne til at optage mere sukker fra blodet og blodsukkerniveauet til at falde.
Med det samme designede AI nye molekyler, der også øger aktiviteten af PPAR'er, ligesom de lægemidler, der er tilgængelige i øjeblikket, men uden en langvarig opdagelsesproces. Efter at ETH-forskerne havde produceret disse molekyler i laboratoriet, udsatte kolleger hos Roche dem for en række forskellige tests. Disse viste, at de nye stoffer faktisk er stabile og ikke-giftige lige fra starten.
Forskerne forfølger nu ikke disse molekyler yderligere med henblik på at bringe lægemidler baseret på dem på markedet. I stedet var formålet med molekylerne at udsætte den nye AI-proces for en indledende streng test.
Schneider siger dog, at algoritmen allerede bliver brugt til lignende undersøgelser på ETH Zürich og i industrien. Et af disse er et projekt med Børnehospitalet Zürich til behandling af medulloblastomer, de mest almindelige maligne hjernetumorer hos børn. Desuden har forskerne offentliggjort algoritmen og dens software, så forskere verden over nu kan bruge dem til deres egne projekter.
"Vores arbejde har gjort en verden af proteiner tilgængelig for generativ AI i lægemiddelforskning," siger Schneider. "Den nye algoritme har et enormt potentiale." Dette gælder især for alle medicinsk relevante proteiner i den menneskelige krop, som ikke interagerer med nogen kendte kemiske forbindelser.
Flere oplysninger: Kenneth Atz et al., Prospective de novo drug design with deep interactome learning, Nature Communications (2024). DOI:10.1038/s41467-024-47613-w
Journaloplysninger: Nature Communications
Leveret af ETH Zürich
 Varme artikler
Varme artikler
-
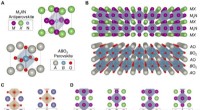 Epitaksiale antiperovskit/perovskite heterostrukturer til materialedesignSkematisk repræsentation af krystalstrukturerne af M3XN nitrid antiperovskite og ABO3 oxid perovskite forbindelser og deres grænseflader. (A) M3XN og ABO3 ideelle enhedsceller, der viser deres geometr
Epitaksiale antiperovskit/perovskite heterostrukturer til materialedesignSkematisk repræsentation af krystalstrukturerne af M3XN nitrid antiperovskite og ABO3 oxid perovskite forbindelser og deres grænseflader. (A) M3XN og ABO3 ideelle enhedsceller, der viser deres geometr -
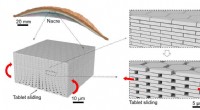 Ny type glas inspireret af naturen er mere modstandsdygtig over for stødMikroarkitekturen af Nacre og tablet, der glider i Nacre. Kredit:Z. Yin, F. Hannard, F. Barthelat Ved at bruge den iriserende perlemor, der ofte findes foring af muslingeskaller, forskere har ko
Ny type glas inspireret af naturen er mere modstandsdygtig over for stødMikroarkitekturen af Nacre og tablet, der glider i Nacre. Kredit:Z. Yin, F. Hannard, F. Barthelat Ved at bruge den iriserende perlemor, der ofte findes foring af muslingeskaller, forskere har ko -
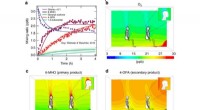 Pig-Pen-effekt:Blanding af hudolie og ozon kan producere en personlig forureningsskyNår ozon rammer olier på huden og i snavset tøj, det kan producere en personlig sky af irriterende stoffer. Kredit:Penn State Når ozon og hudolier mødes, den resulterende reaktion kan hjælpe med a
Pig-Pen-effekt:Blanding af hudolie og ozon kan producere en personlig forureningsskyNår ozon rammer olier på huden og i snavset tøj, det kan producere en personlig sky af irriterende stoffer. Kredit:Penn State Når ozon og hudolier mødes, den resulterende reaktion kan hjælpe med a -
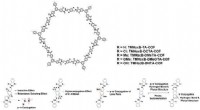 Redigering af lysemitterende organiske molekyler via overflademodifikationFigur 1. Ændringer i lysemission via forstyrrelse af vægoverfladesteder i kovalente organiske rammer (COFer). Ved at introducere forskellige atomer eller små grupper i porestederne af hydrazon-forbund
Redigering af lysemitterende organiske molekyler via overflademodifikationFigur 1. Ændringer i lysemission via forstyrrelse af vægoverfladesteder i kovalente organiske rammer (COFer). Ved at introducere forskellige atomer eller små grupper i porestederne af hydrazon-forbund
- Fed plan:Undersøgelse siger, at bedre klimaanlæg kan bremse den globale opvarmning
- Non-stop signal opnået i højeffekt Erbium-doterede mid-infrarøde lasere
- Den gode, de dårlige og algerne:Salton Havvoksede alger som ny brændstofkilde og forureningsløsni…
- Kontrol af ustabilitet giver et nærmere kig på kemi fra hypersoniske køretøjer
- Ny proces kan reducere energibehovet fra gødning, nitrogenbaserede kemikalier
- Virkningerne af coronavirus på offentlig transport og massetransport