


En dyb variationel autoencoder til proteomik massespektrometri dataanalyse
Jianwei Shuai's team og Jiahuai Hans team på Xiamen University har udviklet en dyb autoencoder-baseret data-uafhængig dataopsamlingsdataanalysesoftware til proteinmassespektrometri, som realiserer analysen af relevante peptider og proteiner fra komplekse proteinmassespektrometridata og demonstrerer overlegenheden og metodens alsidighed på forskellige instrumenter og artsprøver. Undersøgelsen blev offentliggjort i Research som "Kære-DIA XMBD :dyb autoencoder til data-uafhængig opsamlingsproteomik".
Proteiner spiller en central rolle som udfører af cellulære livsaktiviteter og driver et utal af afgørende biologiske processer. Derfor har proteomikområdet fået stor opmærksomhed. Proteomics involverer den omfattende undersøgelse af proteinegenskaber, herunder post-translationelle modifikationer, proteinekspressionsniveauer, protein-protein-interaktioner og mere. Dets overordnede mål er at opnå en holistisk forståelse af sygdomspatogenese, cellulær metabolisme og andre vitale processer på proteinniveau.
Blandt de vigtigste analytiske teknikker inden for proteomikforskning skiller proteinmassespektrometri sig ud som den mest kritiske. Over tid har massespektrometriteknologien udviklet sig til at give forskere pålidelige og dynamiske værktøjer til proteomisk analyse.
To hovedtilgange til proteinmassespektrometri er dataafhængig erhvervelse (DDA) og datauafhængig erhvervelse (DIA). I DDA erhverves alle peptidprecursorionspektre (MS1) i fuld scanningstilstand, efterfulgt af udvælgelse af de mest N-intensive peptidioner til fragmentering for at opnå fragmentionspektre (MS2).
På trods af dets anvendelighed står DDA over for udfordringer relateret til eksperimentel reproducerbarhed og påvisning af peptider med lav overflod på grund af tilfældigheden af peptidfragmentering og den foretrukne udvælgelse af højintensive peptider.
For at overvinde disse begrænsninger er DIA-opsamlingsmetoden blevet introduceret. Denne teknik opdeler masse-til-ladning-forholdsområdet for moderionspektre i flere vinduer og fragmenterer sekventielt alle peptider inden for hvert vindue for at opnå datterionspektre. En almindelig DIA-metode er Sequential Window Acquisition af alle teoretiske fragmentioner (SWATH).
Mens DIA-opsamlingsdata bevarer mere omfattende proteomisk information, udgør dens store datastørrelse, høje dimensionalitet og komplekse spektrale signaler udfordringer for analysen. Som et resultat er DIA-datamining blevet et stort fokus i proteomics-samfundet.
Jianwei Shuai's team og Jiahuai Hans team samarbejdede om at udvikle Dear-DIA, en deep learning-baseret data-uafhængig dataopsamlingsdataanalysesoftware, som realiserer identifikation af fragmentioner svarende til forskellige peptider fra komplekse DIA-opsamlingsspektre og demonstrerer generaliseringen til komplekse prøver fra forskellige arter.
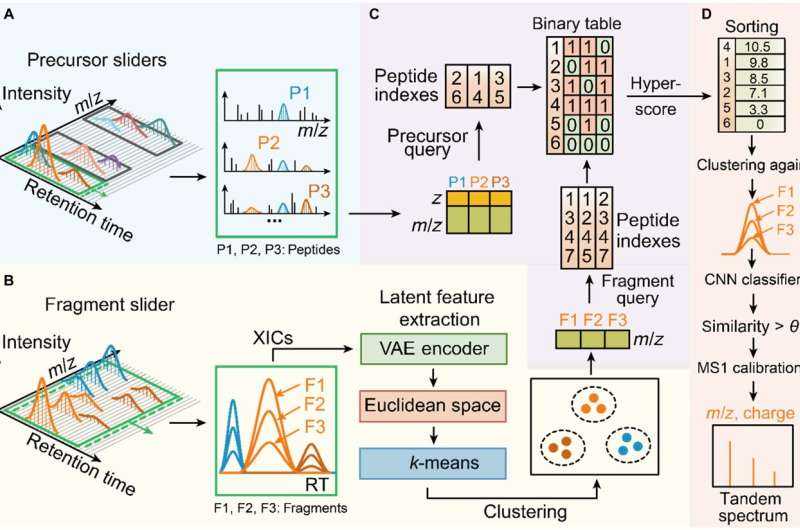
Dear-DIA opdeler først spektrene i en skyder med fast bredde med en fast bredde langs retningen for retentionstid (RT), og hver skyder indeholder et sæt prækursorspektre MS1 og fragmentspektre MS2 som minimumsbehandlingsenhed. Derefter blev en peak-finding-algoritme brugt til at fjerne de lave signal-til-støj-baggrundsioner og fastholde kandidat-precursor-ioner og kandidat-fragment-ioner.
Dernæst bruger Dear-DIA en variationsautoencoder til at udtrække topegenskaberne af fragmentioner og kortlægger funktionerne i det euklidiske rum, og grupperer derefter funktionerne med forskellige klasser af fragmenter, der svarer til forskellige peptider, og realiserer således spektrogram-dekonvolutionsprocessen.
Dear-DIA inkluderer en indekseringsalgoritme kaldet PIndex, som matcher forløberne til fragmenternes klyngeresultater og vælger de bedste parringsresultater ved at score. Dear-DIA bruger et foldet neuralt netværk til at genberegne topformligheden af fragmenter i samme klasse for at eliminere interfererende ioner og klyngeresultater med lav lighed.
Forfatterne testede først ydeevnen af Dear-DIA på et SGS Human-datasæt indeholdende 422 syntetiske peptider af stabile isotopmærkede standarder opdelt i 10 fortyndingsgradienter (fra 1-fold til 512-fold fortynding), og DIA-data blev opnået på en AB SCIEX TTOF5600 massespektrometer, der bruger SWATH-teknikken til at opnå DIA-data.
Analyseresultaterne viste, at Dear-DIA fandt flere syntetiske peptider i alle fortyndede opløsninger sammenlignet med de to almindeligt anvendte analysemetoder, Spectronaut 14 og DIA-Umpire. Forfatterne sammenlignede også antallet af peptider og proteiner fundet ved de forskellige analysemetoder for SGS Human og L929 Mouse datasæt. Resultaterne viste, at Dear-DIA var i stand til at finde flere peptider og proteiner sammenlignet med Spectronaut 14 og DIA-Umpire, hvilket dækkede mere end 85 % af deres resultater.
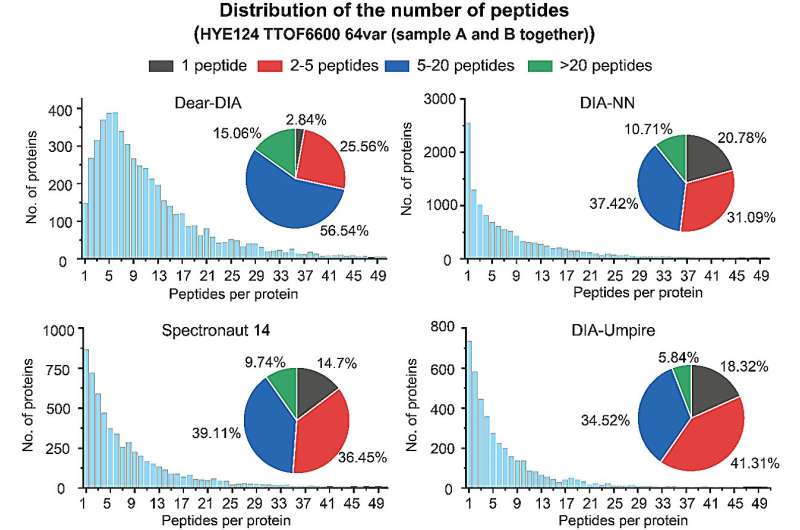
Tilliden til proteomiske analyseresultater kan også demonstreres ved antallet af peptider identificeret for hvert protein. Proteiner med 2 eller flere identificerede peptider anses generelt for at være mere troværdige identifikationer. Forfatterne sammenlignede antallet af proteiner versus peptider rapporteret af Dear-DIA med eksisterende software på et datasæt med blandede arter (HYE124 TTOF6600 64var-datasæt).
Datasættet indeholder proteiner fra tre arter, menneske, gær og E. coli, og dataene blev erhvervet på et AB SCIEX TTOF6600 massespektrometer ved brug af SWATH-metoden, med forældre-ionspektre indeholdende 64 variable vinduer. Analyseresultaterne viste, at 97,16 % af proteinerne fundet af Dear-DIA kunne svare til 2 og flere peptider, hvilket er meget højere end DIA-NN, Spectronaut 14 og DIA-Umpire.
Data-uafhængige indsamlingsteknikker til proteomik er blevet bredt vedtaget, og relaterede analysealgoritmer er blevet et forskningshotspot. Proteinopdagelse fra massive massespektrometridata er en interessant og udfordrende opgave. I denne artikel udviklede holdet Dear-DIA, en analysesoftware baseret på deep learning, som bruges til at behandle en række meget komplekse DIA-opsamlingsdata og kan opdage flere peptider og proteiner, udover at gengive de fleste af resultaterne af Spectronaut og DIA-Umpire.
Selvom træningsdatasættet er fra E. coli, demonstrerer den fremragende ydeevne af Dear-DIA på datasættet med blandede arter dets stærke generaliseringsevne til at analysere komplekse proteomiske data. Deep learning, som et meget brugt værktøj til big data-analyse, har demonstreret fremragende data mining-evner til at opdage dybe iboende associationer i big data.
Brugen af dyb læring til at analysere proteomik-massespektrometridata har et stort potentiale og vil yderligere fremme studiet af grundlæggende spørgsmål såsom proteinsignalnetværk.
Flere oplysninger: Qingzu He et al., Dear-DIA XMBD :Deep Autoencoder muliggør dekonvolution af data-uafhængig opkøbsproteomik, forskning (2023). DOI:10.34133/research.0179
Journaloplysninger: Forskning
Leveret af Research
 Varme artikler
Varme artikler
-
 Godkendelse af hasselnøddernes geografiske oprindelseKredit:American Chemical Society Hasselnødder, som olivenolie, ost og andre landbrugsprodukter varierer i smag afhængigt af deres geografiske oprindelse. Fordi forbrugere og processorer er villige
Godkendelse af hasselnøddernes geografiske oprindelseKredit:American Chemical Society Hasselnødder, som olivenolie, ost og andre landbrugsprodukter varierer i smag afhængigt af deres geografiske oprindelse. Fordi forbrugere og processorer er villige -
 Konstruere et polymernetværk til at fungere som aktiv camouflage efter behovForskernes kunstige kromatoforer består af membraner strakt over cirkulære hulrum fastgjort til pneumatiske pumper. Tryk på hulrummet strækker membranen, ændre tonehøjden for den spiralformede flydend
Konstruere et polymernetværk til at fungere som aktiv camouflage efter behovForskernes kunstige kromatoforer består af membraner strakt over cirkulære hulrum fastgjort til pneumatiske pumper. Tryk på hulrummet strækker membranen, ændre tonehøjden for den spiralformede flydend -
 Udvikling af enzymer til at hjælpe med at løse planetens plastikproblemKredit:Unsplash/CC0 Public Domain Forskere fra Manchester Institute of Biotechnology (MIB) har udviklet en ny enzymteknologisk platform til at forbedre plastnedbrydende enzymer gennem rettet evolut
Udvikling af enzymer til at hjælpe med at løse planetens plastikproblemKredit:Unsplash/CC0 Public Domain Forskere fra Manchester Institute of Biotechnology (MIB) har udviklet en ny enzymteknologisk platform til at forbedre plastnedbrydende enzymer gennem rettet evolut -
 Zinktransportør nøgle til bekæmpelse af kræft i bugspytkirtlen og mereJian Hu og et hold af MSU-forskere har afsløret en nøglestruktur af en molekylær maskine, en ZIP zink transporter. Kredit:MSU Når sporstoffer stiger til giftige niveauer, der sker dårlige ting. P
Zinktransportør nøgle til bekæmpelse af kræft i bugspytkirtlen og mereJian Hu og et hold af MSU-forskere har afsløret en nøglestruktur af en molekylær maskine, en ZIP zink transporter. Kredit:MSU Når sporstoffer stiger til giftige niveauer, der sker dårlige ting. P
- Elon Musk siger, at det ikke er en mulighed at skære ned på arbejdstiden
- Alt vil oprette forbindelse til internettet en dag, og dette biobatteri kunne hjælpe
- Designbehandling af avancerede metaller, der producerer bedre skulptur
- Hvad repræsenterer en kemisk formel?
- En trærig undersøgelse:Biomasse fra skovgenopretning
- Hvordan en partikel kan stå stille i roterende rumtid