


Når molekyler forlader dækspor:En ny tilgang til optimering af molekylær selvorganisering
Visse typer molekyler danner mønstre, når de afsættes på substrater. Fotovoltaiske og sensorenheder fra organiske forbindelser afhænger af dette fænomen med selvorganisering. Fysikere fra Ludwig-Maximilians-Universitaet i München, Tyskland, har nu udviklet en model, der forudsiger disse mønstre og dermed tillader optimering af den molekylære syntese i fremtiden.
Nogle klasser af molekyler er i stand til at arrangere sig selv i specifikke mønstre på overflader. Denne evne til selvorganisering er afgørende for mange teknologiske anvendelser, som er afhængige af samlingen af bestilte strukturer på overflader. Imidlertid, det har hidtil været praktisk talt umuligt at forudsige eller kontrollere resultatet af sådanne processer.
Nu er en gruppe forskere ledet af Dr. Bianca Hermann, en fysiker fra Center for Nanoscience (CeNS) ved LMU München, rapporterer et betydeligt gennembrud:Ved at kombinere statistisk fysik og detaljerede simuleringer med billeder opnået ved scanning tunneling microscopy (STM), holdet har været i stand til at formulere en simpel model, der kan forudsige de observerede mønstre. "Ved hjælp af modellen, vi kan generere en bred vifte af mønstre, der gengiver overraskende godt de arrangementer, der observeres eksperimentelt", siger Hermann. "Vi ønsker at udvide denne tilgang til andre overfladesymmetrier. Allerede nu er områderne for molekylær elektronik, sensor applikationer, overfladekatalyse og organisk fotovoltaik kan drage fordel af vores model. Dens evne til at forudsige strukturer dannet af selvorganisering tillader optimering af molekylære byggesten før syntese." ( Nano bogstaver online, 16. februar 2010)
Når "moder natur" laver ingeniørarbejdet, molekyler kan selvorganisere sig i komplekse strukturer - et første skridt i dannelsen af membraner, celler og andre molekylære systemer. Princippet om selvorganisering, som muliggør en meget økonomisk udnyttelse af ressourcerne, udnyttes også i produktionen af funktionaliserede overflader, der kræves i molekylær elektronik, sensor applikationer, katalyse og fotovoltaiske komponenter. Ideen med fremstillingsprocessen er, at molekylære komponenter bringes i kontakt med et substratmateriale, og derefter "magisk" finde deres foretrukne positioner i det ønskede molekylære netværk. Startkomponenterne er udvalgt til at vise specifikke strukturelle og kemiske egenskaber beregnet til den påtænkte anvendelse. Imidlertid, optimeringen af de molekylære adlayers afhænger i høj grad af en trial-and-error tilgang, og er derfor kompliceret og tidskrævende.
At udvikle den nye model for molekylær-interaktionssted, Dr. Herrmanns gruppe samarbejdede med Priv. Doz. Dr. Thomas Franosch og professor Erwin Frey inden for Cluster of Excellence "Nanosystems Initiative Munich" (NIM). Problemet blev løst ved hjælp af en tilgang fra statistisk fysik kendt som Monte Carlo-metoden, som gør det muligt at udføre en detaljeret computersimulering af statistikken over molekylære interaktioner. De således genererede strukturelle motiver blev sammenlignet med eksperimentelle højopløsningsbilleder af molekylære mønstre opnået ved STM. Marta Balbás Gambra, en doktorand, begyndte hver simulering med en matematisk repræsentation af en samling af hundredvis af tilfældigt orienterede partikler af defineret konformation. Disse skematiske molekyler blev derefter forstyrret ved - beregningsmæssigt - at tilføje energi, får befolkningen til at antage en ny konfiguration.
Ved at bruge denne simuleringsstrategi, man kan generere en større variation af mønstre, end der findes naturligt, og mange af disse svarede nøje til de virkelige molekylære mønstre afsløret af STM. "I et tilfælde forudsagde vi faktisk et mønster, der først senere blev bekræftet med STM", beretter ph.d.-studerende Carsten Rohr. Ifølge termodynamikkens love, fysiske systemer har en tendens til at adoptere den tilstand med den mest gunstige (dvs. laveste) energi. Eksperimentelle tests viste, at forskellige molekylære konfigurationer konverterer indbyrdes, indtil et arrangement dominerer, der minder om dækspor. Og sandelig, Monte Carlo-tilgangen havde forudsagt, at dette arrangement svarer til staten med den laveste energi.
"Til sidst, vi var i stand til at vise, at den molekylære geometri og nogle få fremtrædende træk koder for de observerede strukturelle motiver", forklarer teoretiker Francosch. "Vi planlægger at udvide tilgangen til andre typer overfladesymmetrier, men modellen giver allerede et vigtigt teoretisk værktøj, fordi det hjælper os med at forudsige, hvilken type overflademønster et givet funktionelt molekyle vil danne. Det betyder, at designet af molekyler kan optimeres under den syntetiske fase, for at opnå overflader med de ønskede egenskaber", siger Hermann. Fysikerne i gruppen, som kommer fra forskellige videnskabelige baggrunde og var i stand til at samle deres ekspertise til dette projekt, forestille sig flere potentielle anvendelser for deres model inden for molekylær elektronik, sensorteknologi, katalyse og fotovoltaik. Yderligere muligheder omfatter dets anvendelse til at forudsige resultaterne af andre typer molekylære interaktioner også på delvist mønstrede substrater.
 Varme artikler
Varme artikler
-
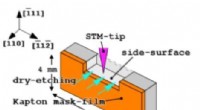 Forskere afbilder perfekt glatte sideoverflader af 3-D siliciumkrystaller med et scanningstunnelmikr…Figur 1. Et skema af en Si(110)-prøve med en Kapton-filmmaske:tør ætsning fra (110) topoverfladen og STM-spids, der nærmer sig (-111) sideoverfladen. Kredit:Osaka University Et forskningssamarbejd
Forskere afbilder perfekt glatte sideoverflader af 3-D siliciumkrystaller med et scanningstunnelmikr…Figur 1. Et skema af en Si(110)-prøve med en Kapton-filmmaske:tør ætsning fra (110) topoverfladen og STM-spids, der nærmer sig (-111) sideoverfladen. Kredit:Osaka University Et forskningssamarbejd -
 Lad der være lys:Kontrolleret oprettelse af kvanteemitterarraysKunstnerens indtryk af enkelte fotoner udsendt fra kvanteprikker i understøttede lagdelte halvledere. Kredit:Pawel Latawiec/Harvard University Overgangsmetal dichalcogenider (TMDer) er lagdelte ha
Lad der være lys:Kontrolleret oprettelse af kvanteemitterarraysKunstnerens indtryk af enkelte fotoner udsendt fra kvanteprikker i understøttede lagdelte halvledere. Kredit:Pawel Latawiec/Harvard University Overgangsmetal dichalcogenider (TMDer) er lagdelte ha -
 Forskere kvantificerer nanopartikel-protein-interaktionerInsulin, et af de mest almindelige proteiner i humant blod, kan akkumulere til fibrøse masser, når den folder forkert. Forskning fra et hold ved NIST viser, at guldnanopartikler tilsyneladende øger in
Forskere kvantificerer nanopartikel-protein-interaktionerInsulin, et af de mest almindelige proteiner i humant blod, kan akkumulere til fibrøse masser, når den folder forkert. Forskning fra et hold ved NIST viser, at guldnanopartikler tilsyneladende øger in -
 Musling-inspireret defektteknologi forbedrer grafenfibrenes mekaniske styrkeTværsnit SEM-billede af ren grafenfiber (venstre) og grafenfiber efter totrinsdefektkontrol ved hjælp af polydopamin (midten og højre). Kredit:KAIST Forskere har demonstreret den muslingeinspirere
Musling-inspireret defektteknologi forbedrer grafenfibrenes mekaniske styrkeTværsnit SEM-billede af ren grafenfiber (venstre) og grafenfiber efter totrinsdefektkontrol ved hjælp af polydopamin (midten og højre). Kredit:KAIST Forskere har demonstreret den muslingeinspirere
- Ny type metasurface tillader hidtil uset laserkontrol
- Fitbit lancerer ny wearable, men din arbejdsgiver skal tilmelde dig
- Indhold mangler i børns bogdelingsoplevelser i lavindkomst, etniske minoritetshusstande
- Forskning i nanopartikler kan forbedre lægemiddellevering gennem huden
- 2015-2016 udløste El Nino sygdomsudbrud over hele kloden
- Undersøgelse afslører ny vej til hurtig, effektiv fjernelse af mikroforurenende stoffer i vand