


Udfoldning af adsorption på metal nanopartikler:Forbindelse af stabilitet med katalyse
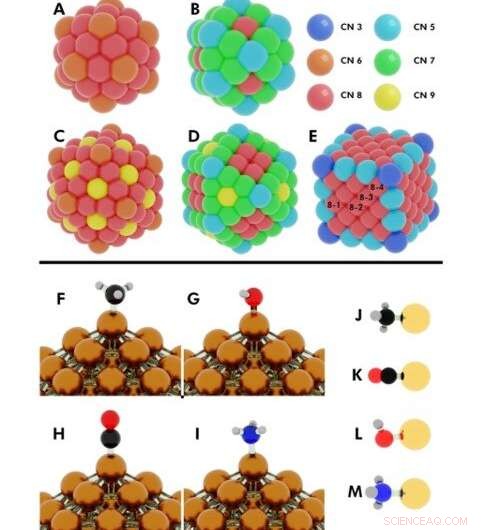
Illustration af de indledende konfigurationer for flere udførte DFT (density functional theory) beregninger. Øvre:Koordinationsnumre på (A) 55-atom icosahedron, (B) 55-atom cuboctahedron, (C) 147-atom icosahedron, (D) 147-atom cuboctahedron, (E) 172-atom terning. Nanopartikler (NP'er), hvor mere end ét unikt atom deler det samme koordinationsnummer (CN), er angivet med tallene 8-1, 8-2, 8-3, 8-4. Kredit:Science Advances, doi:10.1126/sciadv.aax5101
Metal nanopartikler har fået stor opmærksomhed på grund af deres anvendelser inden for forskellige områder fra medicin, katalyse, energi og miljø. Imidlertid, de grundlæggende egenskaber ved nanopartikeladsorption på en overflade mangler at blive forstået. James Dean og et tværfagligt forskerhold i afdelingen for Kemiteknik, i USA indført en universel adsorptionsmodel for at tage højde for de strukturelle egenskaber, metalsammensætning og forskellige adsorbater af nanopartikler via maskinlæring (ML). Modellen passer til et stort antal data for nøjagtigt at forudsige adsorptionstendenser på monometalliske og legeringsbaserede nanopartikler. Skabelonen var enkel og leverede hurtigt beregnede data for metaller og adsorbater. Forskerholdet forbandt adsorptionen med stabilitetsadfærd for at fremme designet af optimale nanopartikler til applikationer af interesse. Forskningen er nu offentliggjort på Videnskabens fremskridt .
Metal nanopartikler (NP'er) har betydelige anvendelser inden for katalyse, lige fra brændstof- og kemikalieproduktion, til solenergi og kemisk energi. Men deres stabilitet og katalytiske aktivitet viser generelt modsatrettede tendenser, hvor meget aktive katalysatorer kun kan fungere i nogle få cykler. Et nøgletræk ved omfanget af metallisk katalytisk funktionalitet afhænger af adsorptionsstyrken for en række arter på katalysatoroverfladen. Ifølge Sabatier-princippet, udviklet for mere end et århundrede siden, aktive katalysatorer bør binde adsorbater med en bindingsstyrke, der hverken er stærk eller svag. Mens stærkt adsorberede arter kan forgifte katalysatoroverfladen, svagt bundne reaktanter desorberes let. I et mellemscenarie, reaktanterne kan møde hinanden og reagere på de katalytiske overflader. Forskere bruger i øjeblikket beregningssimulering og teoretiske kemimetoder til at stimulere katalytisk adfærd på metalkatalysatorer med stor nøjagtighed til at guide efterfølgende eksperimenter i laboratoriet.
Beregningsmæssige bestræbelser har fokuseret på at screene forskellige metalkatalysatorer for at opdage den "magiske" bindingsenergi (BE) af kemiske arter på katalysatoroverflader for at danne meget aktive katalysatorer. In silico design af katalytisk aktive materialer, imidlertid, mangler at blive realiseret. Ulemperne skyldes hovedsageligt designbestræbelser, der ofte forsømmer katalysatorernes stabilitet. NP-katalysatorer har også en høj grad af site-heterogenitet på deres overflade til adsorption og katalyse. Forskere havde udviklet adsorptionsmodeller til at relatere bindingsenergi af adsorbaterne til overfladekarakteristika for NP'er, såsom koordinationsnumre (CN'er) for at forstå det stedspecifikke adsorptionsrespons. Endnu, for klarhed, variationen i bindingsenergi (BE) medfører også sekundære deskriptorer såsom krumning og elektroniske egenskaber af NP'er.
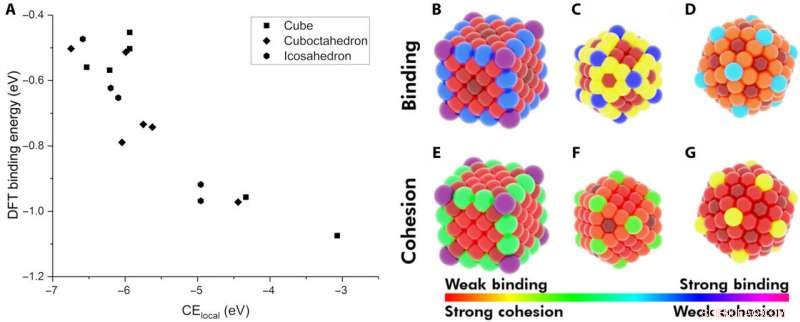
Demonstration af lokal sammenhængende energi (CElocal) som en deskriptor for adsorptionsenergi. (A) BE for CO på forskellige steder i Au NP'er som funktion af CElocal:172-atom-terning (rektangler), 147-atom icosahedron (sekskanter), og 147-atom cuboctahedron (rhombus). Varmekort over forskellige steder på NP'erne med hensyn til deres BE af CO (B til D) og til deres CElokale (E til G). Farveskemaet følger rækken af stærkeste CO-binding til svageste CElocal (violet) og af svageste binding til stærkeste CElocal (rød). Kredit:Science Advances, doi:10.1126/sciadv.aax5101
I nærværende arbejde, Dean et al. anvendt tæthedsfunktionel teori (DFT) og maskinlæringsteknikker til at udlede en simpel fysikbaseret model til nøjagtigt at fange den varierende adsorptionsenergi. De estimerede variablen som en funktion af det lokale adsorptionsstedsmiljø på NP-overfladen såvel som typen af metal-NP. Den generaliserede model kunne anvendes på enhver metalnanostruktur for at forstå adsorptionsadfærd på NP-katalysatoren og katalysatorens stabilitet; at screene og designe katalysatorer til adskillige applikationer.
Forskerne antog først de vigtigste faktorer mellem monometalliske NP'er og adsorbater. De definerede derefter den lokale sammenhængende energi (CE lokal ) i bulkmetaller og opfangede CE'er i NP'er ved hjælp af en bindingscentreret model, som opsummerede hver metal-metal bindingsenergi. By applying similar concepts, they described the stability of binding sites and showed how chemically unsaturated sites (fewer metal-metal bonds) bound adsorbates with an increased strength. The research team focused on describing the binding capacity of a single adsorbate-metal pair. They plotted the DFT-calculated binding energy of carbon monoxide (CO) to a 172-atom gold (Au) cube and a 147-atom gold (Au) cuboctahedron or icosahedron. The team observed a strongly inverse relationship between the local cohesive energy (CE lokal ) and binding energy (BE) to suggest the strongest adsorption sites to be those exhibiting the weakest local cohesion.
The team further developed their model and performed ordinary least squares (OLS) regression to understand adsorption on monometallic NPs and slabs using three adsorbates [Methyl radical (CH 3 ), CO, hydroxyl radical (OH)] on three different metals (Cu, Ag—silver, Au). The metallic NPs contained different morphologies (172-atom cube, 55- and 147-atom icosahedron and 55- and 147-atom cuboctahedron). They observed that the binding affinity to the adsorbates decreased as the cohesion of the local sites increased. And as the adsorbate's chemical potential increased, they became less stable and bound a metal NP with higher tendency. The direct correlation with the metal Adsorbate (MAD) intuitively described the tendency of the metal to bind the adsorbate.

Parity plot of the model-predicted binding energy (BE) of adsorbates (OH, CO, and CH3) on various metal systems versus the DFT BE (eV). (A) The model both trained and tested on PBE DFT data for NPs (Au/Ag/Cu, 55 to 172 atoms), which includes the nanoparticle cohesive energy (CENP) term. (B) The model both trained and tested on PBE DFT data for NPs (Au/Ag/Cu, 55 to 172 atoms), which does not include the CENP term. (C) The model trained on PBE DFT data for NPs (Au/Ag/Cu, 55 to 172 atoms) and tested against RPBE (revised Perdew-Burke-Ernzerhof model) DFT data for top-site adsorptions on metal surfaces (Au/Ag/Cu). (D) The model both trained and tested on RPBE DFT data for top-site adsorptions on metal surfaces (Au/Ag/Cu) from the slab dataset. Kredit:Science Advances, doi:10.1126/sciadv.aax5101
Dean et al. tested the generalizability of the model and trained the simulation on a single metal or single morphology, although it accurately captured other metals or morphologies as well. The work provided strong evidence that the model captured the underlying physics of the binding interactions, allowing the team to extend the work from non-periodic NPs to periodic slab systems. Computationally inexpensive systems could parameterize the model to extend to larger systems, which was not thus far possible due to the computational costs involved.
Dean et al. then extended the model from monometallic NPs to bimetallic systems. Til disse eksperimenter, they plotted the BE—trained on monometallic NPs, across several sites of bi-metallic, 55-atom icosahedron NPs (Cu 31 Ag 24 and Cu 22 Ag 33 ). The model very accurately captured trends in adsorption on the bimetallic Cu/Ag NPs as well. This was an interesting result since the scientists had only trained the model on monometallic systems. The results showed the generalizability of the model for both monometallic and bimetallic NPs. Imidlertid, the team will account additional descriptors including binding site electronegativity to understand the adsorption behavior for bimetallic systems in depth.
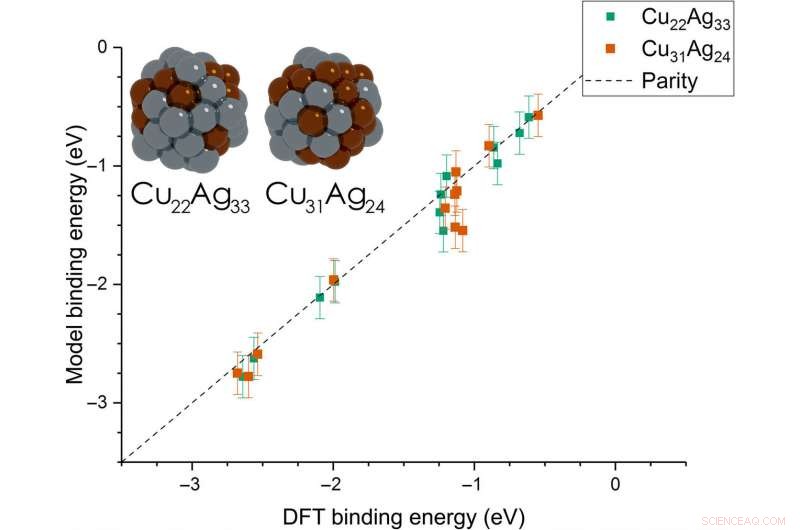
Parity plot between the presently developed model and DFT calculations on icosahedral bimetallic (Cu55−xAgx, x =24, 33) NPs. The model is trained on CH3, CO, and OH adsorbing on monometallic Ag, Cu, and Au NPs and is able to capture adsorption on bimetallic NPs. Images of the two NPs are shown as inset, with copper and silver atoms colored in brown and gray, henholdsvis. Kredit:Science Advances, doi:10.1126/sciadv.aax5101
Although Dean et al. trained the ML (machine learning) algorithm to capture the adsorption trends of just one type of d9 metal, it could accurately predict the behavior of similar d9 metals (Cu—coper, Ag and Au). When they trained the model on a dataset of CH 3, CO and OH adsorbed to Cu, Ag and Au NPs, they could also capture general adsorption trends for similar elements in other columns of the periodic table. They then improved the complexity of the machine learning techniques to provide additional avenues to improve the model of adsorption.
På denne måde James Dean and his colleagues developed a simple yet powerful physics-based model to capture trends on the strength of binding interactions between different adsorbates and metal NPs using machine learning techniques. The study was the first to develop an adsorption model that accurately connected the properties of diverse metal NPs with the stability of the adsorption site. The model introduced simple descriptors to capture the adsorption on any site, relative to monometallic and bimetallic NPs. The team generalized the model to effectively stimulate a range of binding interactions, including variations on the types of metals, their composition, sites of adsorption and adsorbates.
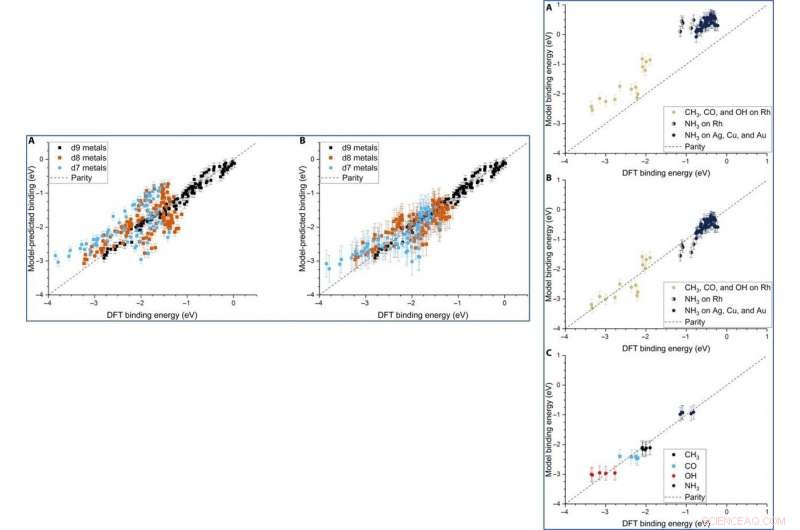
LEFT:The three-descriptor model extended to slab dataset. (A) The model trained on the slab dataset on Cu, Ag, and Au surfaces and tested against the Rh, Ir, Ni, Pd, Pt, Cu, Ag, and Au surfaces from the slab dataset. (B) The equivalent model when trained separately for each column of the d-block, still using the slab dataset. Error bars in every case are the 10-fold cross-validated RMSE of the training set. RIGHT:Extension of the model to Rh and NH3. (A) The model parameterized on our Ag, Cu, and Au NPs adsorbing CH3, CO, and OH and tested against Rh and NH3. (B) The equivalent model with empirical (constant) corrections for Rh and NH3. In the case of NH3 bound to Rh, both corrections are simultaneously applied and indicated by two-colored dots. (C) The model trained on CH3, CO, OH, and NH3 adsorbing on icosahedral/cuboctahedral Rh55. Kredit:Science Advances, doi:10.1126/sciadv.aax5101
Although the team did not test the applicability of the model for ternary systems, the physical properties may remain relevant to accurately model multimetallic systems as well. The adsorption model can accurately describe the binding strength of a variety of molecules on any site of NPs, including alloys. The scientists expect the model to be highly applicable as a screening tool for the high throughput search of potential catalysts.
© 2019 Science X Network
Sidste artikelGrafen er 3-D såvel som 2-D
Næste artikelPlasmoniske sølv-nanopartikler går frem mod ultrahurtig detektion af enkeltmolekyler
 Varme artikler
Varme artikler
-
 En revolution i lyset i den lille skalaEt kunstnerisk syn på en magnetisk dipolresonans i en dielektrisk kugle med højt brydningsindeks. Kredit:Genoptrykt med tilladelse fra AAAS Lys opfører sig på ret tamme og forudsigelige måder, når
En revolution i lyset i den lille skalaEt kunstnerisk syn på en magnetisk dipolresonans i en dielektrisk kugle med højt brydningsindeks. Kredit:Genoptrykt med tilladelse fra AAAS Lys opfører sig på ret tamme og forudsigelige måder, når -
 Stammeinduceret isomerisering af molekylære kæderFigur (A) giver en skematisk illustration af syntesen af metal-organiske kæder (MOCer) og deres strukturelle afslapning på et kobbersubstrat. Forbindelse 1, 5-dibromo-2, 6-dimethylnaphthalen (DBDMN)
Stammeinduceret isomerisering af molekylære kæderFigur (A) giver en skematisk illustration af syntesen af metal-organiske kæder (MOCer) og deres strukturelle afslapning på et kobbersubstrat. Forbindelse 1, 5-dibromo-2, 6-dimethylnaphthalen (DBDMN) -
 Inkorporerer ferromagnetisme og superledning i et enkelt lag af molekylært supergitterFigur (A) giver en skematisk illustration af ICCD-metoden (Interlayer-space confined chemical design) til syntese af tantaldisulfid (TaS) 2 ) molekylært supergitter med de superledende områder og fe
Inkorporerer ferromagnetisme og superledning i et enkelt lag af molekylært supergitterFigur (A) giver en skematisk illustration af ICCD-metoden (Interlayer-space confined chemical design) til syntese af tantaldisulfid (TaS) 2 ) molekylært supergitter med de superledende områder og fe -
 Ny teknologi kan øge hastigheden og følsomheden for sygdomsdetekteringstest (m/ video)Ayse Rezzan Kose (billedet) og Hur Koser fra Yale University udviklede en ny metode til at identificere og sortere syge celler i blodprøver ved hjælp af de magnetiske nanopartikler i ferrofluider. Kre
Ny teknologi kan øge hastigheden og følsomheden for sygdomsdetekteringstest (m/ video)Ayse Rezzan Kose (billedet) og Hur Koser fra Yale University udviklede en ny metode til at identificere og sortere syge celler i blodprøver ved hjælp af de magnetiske nanopartikler i ferrofluider. Kre
- Oprettelse af selvkonstruerede foldede makrocykler med lav symmetri
- Forskere finder en måde at skrælle slimede biofilm som gamle klistermærker
- Ny forskning fremhæver virkningen af COVID-19 på fødevaresikkerheden i Kenya og Uganda
- Alkoholer som kulstofradikale forstadier
- Reaktion på brand påvirker vandstanden 40 år ud i fremtiden
- Et stykke Mars skal hjem