


Forstå grænseflader mellem hybride materialer og maskinlæring
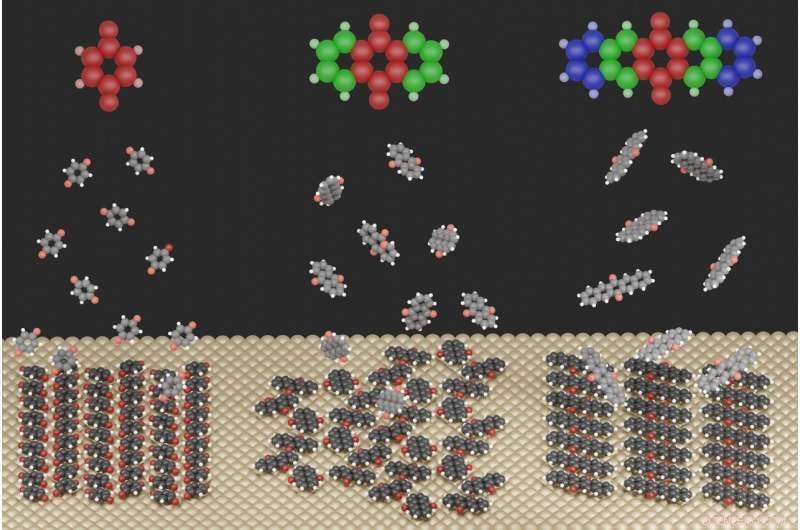
Illustrationen viser de stærkt forskellige overfladestrukturer, der dannes for de tre undersøgte molekyler, når de adsorberes på en metaloverflade. Kredit:Jeindl—TU Graz
Brug af maskinlæringsmetoder, forskere ved TU Graz kan forudsige strukturdannelsen af funktionaliserede molekyler ved grænsefladerne mellem hybridmaterialer. Nu er det også lykkedes at se bag om drivkræfterne i denne strukturdannelse.
Produktionen af nanomaterialer involverer selvsamlingsprocesser af funktionaliserede (organiske) molekyler på uorganiske overflader. Denne kombination af organiske og uorganiske komponenter er afgørende for anvendelser inden for organisk elektronik og andre områder af nanoteknologi.
Indtil nu, visse ønskede overfladeegenskaber blev ofte opnået på en trial-and-error basis. Molekyler blev kemisk modificeret, indtil det bedste resultat for den ønskede overfladeegenskab blev fundet. Imidlertid, de processer, der styrer selvsamlingen af molekyler ved grænseflader, er så komplekse, at små molekylære ændringer kan føre til helt andre motiver. Fysikere fra TU Graz forklarer denne uventede strukturdannelse i en undersøgelse offentliggjort i det anerkendte tidsskrift ACS Nano .
Til dette formål, forskerne studerede quinoidforbindelser på en sølvoverflade. Første forfatter Andreas Jeindl fra Institute of Solid State Physics forklarer:"Naivt, man kunne forvente, at molekyler med lidt forskellige størrelser, men den samme funktionalisering, danner lignende motiver. I slående kontrast, vores fælles teoretiske og eksperimentelle undersøgelse viser, at quinoner kan danne forskellige strukturer. På trods af konstante startbetingelser, dannelsen af disse strukturer kan ikke forudsiges og planlægges uden detaljeret viden om de relevante interaktioner."
Tre modsatrettede drivkræfter
Forskerne i Graz, sammen med et hold fra FSU Jena, er nu begyndt at nedbryde denne uforudsigelighed. De fandt ud af, at strukturdannelsen er resultatet af en afvejning mellem tre modsatrettede drivkræfter:Interaktionen mellem molekyler og metallet forsøger at tvinge alle molekyler i samme orientering, mens interaktionen mellem molekyler nogle gange favoriserer forskellige orienteringer. Molekylernes geometriske former fungerer så som en tredje faktor, forhindrer eller kun delvist tillader visse interaktioner.
Baseret på dette, de var i stand til at etablere et designprincip, hvormed de strukturer, der dannes ved grænsefladerne, og efterfølgende deres egenskaber, kan forudsiges - i det mindste for en første klasse af molekyler. En essentiel rolle spilles af en søgealgoritme (SAMPLE) baseret på maskinlæring. Jeindl uddyber:"Vi var i stand til at vise i denne publikation, at de strukturer, der forudsiges af vores algoritme, er i fremragende overensstemmelse med eksperimentelle karakteriseringer af organisk-uorganiske grænseflader - både i hvordan molekylerne orienterer sig på overfladen og i hvordan motiverne gentages på overflade. Desuden vores analyse, for første gang, muliggjorde en detaljeret og kvantitativ opdeling af drivkræfterne, ikke kun af de eksperimentelt dannede strukturer, men de facto af alle tænkelige strukturer. Dette er et vigtigt kig bag kulisserne af strukturdannelse."
Grænsefladeegenskaber med modulære byggeklodser
Det ikke-intuitive samspil mellem lignende vigtige interaktionsmekanismer er fortsat en udfordring for design af funktionelle grænseflader. Med en detaljeret undersøgelse af alle drivkræfterne, imidlertid, fysikerne ved TU Graz er ikke desto mindre i stand til at udtænke et designprincip for selvsamling af funktionaliserede molekyler for en given klasse af molekyler. Når først der er nok analyser for forskellige klasser af molekyler, de rigtige molekyler til de ønskede grænsefladeegenskaber kan nemt samles på computeren fra modulære byggeklodser.
Sidste artikelForskere er nu i stand til at kortlægge defekter i 2D-krystaller i væske
Næste artikelSporing af topologiske forhold i grafen
 Varme artikler
Varme artikler
-
 Forskere udvikler tre-trins proces til at bygge fraktale nanostrukturerFancy erektorsæt? Nix. Den omfattende fraktale struktur vist til højre (med et nærbillede nedenfor) er mange, mange gange mindre end det og er bestemt ikke en barneleg. Det er det seneste eksempel på,
Forskere udvikler tre-trins proces til at bygge fraktale nanostrukturerFancy erektorsæt? Nix. Den omfattende fraktale struktur vist til højre (med et nærbillede nedenfor) er mange, mange gange mindre end det og er bestemt ikke en barneleg. Det er det seneste eksempel på, -
 Strækbar, transparent grafen-metal nanotrådselektrodeDette er en LED-monteret blød øjenkontaktlinse. Kredit:UNIST En hybrid gennemsigtig og strækbar elektrode kunne åbne den nye vej for fleksible skærme, solceller, og endda elektroniske enheder mont
Strækbar, transparent grafen-metal nanotrådselektrodeDette er en LED-monteret blød øjenkontaktlinse. Kredit:UNIST En hybrid gennemsigtig og strækbar elektrode kunne åbne den nye vej for fleksible skærme, solceller, og endda elektroniske enheder mont -
 Låser op for rigere intracellulære optagelserSEM-billeder giver et nærmere kig på 3DFG-elektroder. Kredit:Carnegie Mellon University, Institut for Biomedicinsk Teknik Bag hvert hjerteslag og hjernesignal er et massivt orkester af elektrisk a
Låser op for rigere intracellulære optagelserSEM-billeder giver et nærmere kig på 3DFG-elektroder. Kredit:Carnegie Mellon University, Institut for Biomedicinsk Teknik Bag hvert hjerteslag og hjernesignal er et massivt orkester af elektrisk a -
 Gennembrud inden for molekylær elektronik baner vejen for DNA-baserede computerkredsløb i fremtide…Baner vejen for en ny generation af DNA-baserede computerkredsløb:Prof. Danny Porath, fra Hebraisk Universitets Institut for Kemi og Center for Nanovidenskab og Nanoteknologi. Kredit:Hebrew University
Gennembrud inden for molekylær elektronik baner vejen for DNA-baserede computerkredsløb i fremtide…Baner vejen for en ny generation af DNA-baserede computerkredsløb:Prof. Danny Porath, fra Hebraisk Universitets Institut for Kemi og Center for Nanovidenskab og Nanoteknologi. Kredit:Hebrew University
- Afslører kompleks adfærd af en turbulent fane ved den kælvende front af en grønlandsk gletsjer
- Kinds of Reasoning in Geometry
- Kvinder er mindre tilbøjelige til at udvikle sig på arbejdet end deres mandlige kolleger efter fø…
- Billede:Hubble tager et portræt af den forsvundne galakse
- Massiv engelsk retssag over VW dieselgate når retten
- Mål reducerer leveringsgebyret med næsten det halve, lægge pres på Walmart, Amazon