


En ny frastødningsmodel for grafenkatalysatorer
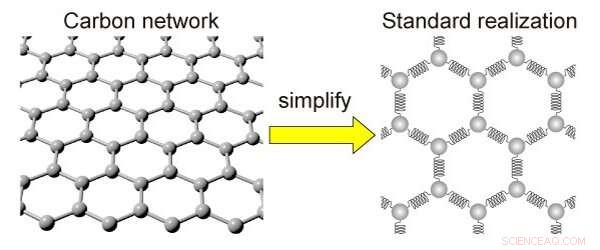
Forenklingen af et kulstofnetværk. Kulstofnettet kan udskiftes med kugler og fjeder for forenkling. Kredit:Kotani et al
En ny matematisk model hjælper med at forudsige de små ændringer i kulstofbaserede materialer, der kan give interessante egenskaber.
Forskere ved Tohoku University og kolleger i Japan har udviklet en matematisk model, der abstraherer de vigtigste virkninger af ændringer i kulstofmaterialets geometrier og forudsiger dets unikke egenskaber.
Detaljerne blev offentliggjort i tidsskriftet Kulstof .
Forskere bruger generelt matematiske modeller til at forudsige de egenskaber, der kan opstå, når et materiale ændres på bestemte måder. Ændring af geometrien af tredimensionel (3D) grafen, som er lavet af netværk af kulstofatomer, ved at tilføje kemikalier eller indføre topologiske defekter, kan forbedre dets katalytiske egenskaber, for eksempel. Men det har været svært for videnskabsmænd at forstå, hvorfor det netop sker.
Den nye matematiske model, kaldet standardrealisering med frastødende interaktion (SRRI), afslører forholdet mellem disse ændringer og de egenskaber, der opstår ved dem. Det gør dette ved at bruge mindre regnekraft end den typiske model, der anvendes til dette formål, kaldet density functional theory (DFT), men det er mindre nøjagtigt.
Med SRRI-modellen, forskerne har forfinet en anden eksisterende model ved at vise de tiltrækkende og frastødende kræfter, der eksisterer mellem tilstødende atomer i kulstofbaserede materialer. SRRI-modellen tager også højde for to typer krumning i sådanne materialer:lokale krumninger og middel krumning.
Forskerne, ledet af Tohoku University matematiker Motoko Kotani, brugte deres model til at forudsige de katalytiske egenskaber, der ville opstå, når lokale krumninger og dopingmidler blev introduceret i 3D-grafen. Deres resultater lignede dem, der blev produceret af DFT-modellen.
"Nøjagtigheden af SRRI-modellen viste en kvalitativ overensstemmelse med DFT-beregninger, og er i stand til at screene gennem potentielle materialer omkring en milliard gange hurtigere end DFT, " siger Kotani.
Holdet fremstillede derefter materialet og bestemte dets egenskaber ved hjælp af scanning elektrokemisk cellemikroskopi. Denne metode kan vise en direkte sammenhæng mellem materialets geometri og dets katalytiske aktivitet. Det afslørede, at de katalytisk aktive steder er på de lokale krumninger.
"Vores matematiske model kan bruges som et effektivt præ-screeningsværktøj til at udforske nye 2-D- og 3D-kulstofmaterialer for unikke egenskaber før anvendelse af DFT-modellering, " siger Kotani. "Dette viser vigtigheden af matematik for at accelerere materialedesign."
Holdet planlægger derefter at bruge deres model til at lede efter forbindelser mellem designet af et materiale og dets mekaniske og elektrontransportegenskaber.
 Varme artikler
Varme artikler
-
 Miljøvenlige fotoluminescerende nanopartikler til mere levende displayfarverFig. 1. Fotoluminescensspektre af iridiumkompleks og konventionelle cadmiumselenid -kvantepunkter. Kredit:Osaka University Forskere på universitetet ledet af Osaka skabte en ny type lysemitterende
Miljøvenlige fotoluminescerende nanopartikler til mere levende displayfarverFig. 1. Fotoluminescensspektre af iridiumkompleks og konventionelle cadmiumselenid -kvantepunkter. Kredit:Osaka University Forskere på universitetet ledet af Osaka skabte en ny type lysemitterende -
 Forskere demonstrerer praktiske metal nanostrukturerEn kunstners syn på en metasflade bestående af en rektangulær række af rektangulære guldnanostrukturer, der genererer plasmoniske overfladegitterresonanser. Kredit:Illustration af Yaryna Mamchur, medf
Forskere demonstrerer praktiske metal nanostrukturerEn kunstners syn på en metasflade bestående af en rektangulær række af rektangulære guldnanostrukturer, der genererer plasmoniske overfladegitterresonanser. Kredit:Illustration af Yaryna Mamchur, medf -
 Blanding af grafen nanobånd, polymer har potentiale for biler, soda, ølEt sammensat materiale, der er skabt ved Rice University, er næsten uigennemtrængeligt for gas og kan føre til effektiv opbevaring af komprimeret naturgas til køretøjer. En 65 mikrometer bred polymerf
Blanding af grafen nanobånd, polymer har potentiale for biler, soda, ølEt sammensat materiale, der er skabt ved Rice University, er næsten uigennemtrængeligt for gas og kan føre til effektiv opbevaring af komprimeret naturgas til køretøjer. En 65 mikrometer bred polymerf -
 Ny indsigt i memristive enheder ved at kombinere begyndende ferroelektrik og grafenDenne illustration viser, hvordan strontiumtitaniumoxid kombineres med grafenstrimler. Kombinationen åbner en ny vej til memristive heterostrukturer, der kombinerer ferroelektriske materialer og 2D-ma
Ny indsigt i memristive enheder ved at kombinere begyndende ferroelektrik og grafenDenne illustration viser, hvordan strontiumtitaniumoxid kombineres med grafenstrimler. Kombinationen åbner en ny vej til memristive heterostrukturer, der kombinerer ferroelektriske materialer og 2D-ma
- Den første beboelige zone, Planet på størrelse med jorden opdaget med exoplanetundersøgelsesrumf…
- Sådan finder du hvor mange mol der er i en forbindelse
- Sådan beregnes vægten af vinkel Iron
- Forskere bruger nye datavidenskabelige værktøjer til at fange enkeltmolekyler i aktion
- Forskere udfører de første in situ-strålingsmålinger 21 km i luften over det tibetanske plateau
- Hubble sætter fokus på en himmelsk sidemand